
Meeting Summary
Prof Augustin opened the meeting and reviewed the use of systemic treatments recommended for moderate-to-severe psoriasis. Data indicating a lack of access to biologic treatments were presented, and barriers to the use of biologics, including both patient and physician-related cost, were discussed. The opportunity for improved access to biologic treatment options, portrayed by the availability of biosimilars, and the potential to improve healthcare for patients with psoriasis was presented.
Dr Schiestl explained that the demonstration of biosimilarity for regulatory requirements is based on the totality of evidence generated from analytical, non-clinical, and clinical data. The physicochemical and biological assessments performed for comparison of the proposed biosimilar and the originator molecule, using state-of-the-art technology, are most sensitive. Comparative, analytical, and functional testing therefore represent the major part of the comparability exercise, proving that the biosimilar and originator product contain essentially the same active substance. After demonstration of similarity at an analytical and functional level, suitable comparative pharmacodynamics (PDs) and/or pharmacokinetics (PKs) and/or safety studies in animal models are performed. Comparative clinical PKs/PDs and safety is assessed in healthy volunteers as an essential part of the clinical development programme. A final confirmatory Phase III clinical study is conducted in a sensitive patient population to confirm similar safety and efficacy of the biosimilar compared to the originator molecule.
Dr Gerdes explained why psoriasis is a sensitive and robust indication for confirming clinical efficacy of a biosimilar. He presented data from the EGALITY confirmatory study of the etanercept biosimilar (GP2015) in patients with moderate-to-severe psoriasis to compare safety and efficacy, and provided data on multiple switches between the originator etanercept (ETN) and the biosimilar. The trial confirmed the clinical equivalence of the efficacy and safety of GP2015 with ETN; no new safety signals were observed. Switching between the originator and biosimilar had no effect on safety or efficacy over the 52-week study.
The Evolving Role of Systemic Therapy in Psoriasis
Professor Matthias Augustin
Psoriasis is a key disease for dermatologists, with a prevalence in Europe of 2.5% (approximately 15 million people), with 20–25% of these having severe disease. Recent European and World Health Organization (WHO) reports1,2 have recommended that patients should have early, comprehensive treatment, and have highlighted costs limiting access to biologics as the main barrier. Healthcare systems also affect access to biologics for psoriasis. The European Dermatology Survey found that the prescription of systemic psoriasis treatment by dermatologists varies between countries and in some European countries biologic treatments can only be prescribed by hospitals. Around half of European countries have fixed budgets for prescribing biologics in psoriasis, and two-thirds require additional approval of the clinician’s prescription. The importance of treatment access is demonstrated by the significant link between lack of access to multiple systemic treatments and reduced treatment success.3
Over the last decade, systemic biologic therapies for psoriasis have become widespread, with new biologics and biosimilars of existing biologics anticipated within the next few years. The challenge for dermatologists is how best to use this abundance of new treatments. The patient needs which dermatologists are addressing are wider than simply improvements in skin conditions, as measured by the Psoriasis Area and Severity Index (PASI).4 Current European guidelines5,6 recommend several systemic biologic treatments for moderate-to-severe psoriasis, as well as conventional systemic therapies and ultraviolet (UV) treatment, but there is currently no clear guidance on which systemic therapy to use first, nor on switching biologics. The selection of a systemic treatment for psoriasis should take into account the clinical criteria, the drug characteristics, the patient characteristics, and the regulatory requirements (including national healthcare system regulations). Treatment goals for moderate-to-severe psoriasis have been defined, and the European consensus is that the ultimate goal of any treatment is the complete clearance of lesions.7 However, while the complete clearance of lesions (PASI score 90–100) is a good indicator of drug performance, the most patient-relevant endpoints are actually PASI 50, 75, and 90 scores.8 Early intervention with systemic treatment provides benefit in terms of minimising increases in morbidity and patient suffering, and in minimising associated costs.9 The long-term safety of treatment is also a key factor as psoriasis is a lifelong disease. Data from pharmacovigilance registries are essential for patient safety, as patients in the real world can have different rates of adverse events to those selected for clinical trials.10 Data from the PsoBest registry show that the rate of serious adverse events is similar between conventional systemic treatments for psoriasis and biologics.11
Despite the guidelines and evidence for both benefit and long-term safety, many patients with severe disease still do not get access to systemic therapy.12 These barriers to guideline-compliant psoriasis care come from patients, physicians, and external factors. Patients are unaware of the benefits of systemic therapies and have misconceptions of the risks, whilst physicians can lack knowledge of the guidelines or have a fear of legal liability in the case of adverse outcomes. External factors such as poor healthcare infrastructure, low budgets, or lack of training may also prevent guideline-compliant psoriasis care.13 Recommendations have been made to overcome these barriers, and these include acknowledging the financial implications of treating a complex disease such as severe psoriasis.13
The increasing number of biologics and biosimilars in the market and in development provides the opportunity to overcome the cost barrier in accessing systemic treatment for psoriasis. The use of biosimilars has been shown to reduce healthcare costs,14 and the estimated cost savings achieved by use of an ETN biosimilar instead of ETN (between 2016 and 2020) could provide the opportunity to fund treatment for an additional 3,100 patients in the UK or 17,310 patients in Germany.15 Therefore, improved access and lower costs of systemic therapy have the potential to allow innovation in the treatment paradigm for patients with psoriasis, as well as enabling a more patient-centred approach to treatment.16-18
In summary, biologic treatments have changed the landscape of psoriasis care and provide added value. There is clear evidence for starting systemic treatment early, but with the variety of effective systemic treatment options for moderate-to-severe psoriasis there is no clear evidence about optimum treatment pathways or long-term management. Choices for systemic treatment take into account clinical characteristics of the patient’s psoriasis, drug properties, and patient preferences. There is a large body of evidence generated by real-world safety data of systemic biologic treatments for psoriasis, but other barriers to their use exist. Removing these barriers will involve creating awareness of the evidence for their benefits, disseminating expert knowledge of their use, optimising prices, and making use of the potential of biosimilars.
Analytical Comparison as the Foundation of Biosimilarity
Doctor Martin Schiestl
Despite biosimilars having been approved for use within Europe for the past 10 years, there is still a lack of awareness regarding their development. The essential point is the paradigm shift involved; in contrast to the development of an originator where the major goal is to determine the clinical efficacy and safety, the major goal in biosimilar development is to demonstrate that the biosimilar and originator are structurally and functionally comparable. Analytical methods provide the most sensitive tools to establish head-to-head comparability and thus provide the foundation of biosimilar development.19 Tailored head-to-head non-clinical and clinical studies are focussed on identifying any differences between the originator and the biosimilar, if they exist, rather than re-establishing the clinical efficacy and safety de novo, which has already been demonstrated by clinical studies of the originator. In Europe and the USA, the term ‘biosimilar’ is strictly defined as a biologic product (usually a successor to a biologic that has lost exclusivity) that has been approved via a stringent regulatory pathway. An approved biosimilar matches the reference biologic, and patients and physicians can expect the same safety and efficacy profile.20
An approved biosimilar must match its reference biologic structurally. The amino acid sequence has to be identical and the folding that results in the three-dimensional structure (the secondary, tertiary, and quaternary structure of the protein) must be indistinguishable from the originator.21 Post-translational modifications of the protein such as glycation, which are highly variable in nature, must contain identical structures in comparable amounts. Differences in these are only acceptable if it can be demonstrated that they do not lead to any clinically relevant effects.21 A degree of variability is natural in glycated proteins, and batch-to-batch variability in glycation can readily be measured in commercially available complex biological products e.g. monoclonal antibodies (Figure 1). Changes in the manufacturing process, such as implementation of a new purification method or a change in manufacturing site, may also cause minor but measurable changes in the glycation or the purity profile of a biologic.22 These changes are stringently controlled by regulators and are only approved if they do not lead to clinically meaningful differences. Companies may change manufacturing processes for originator biologics many times after approval,23 and these changes are well understood and tightly controlled by health authorities. The scientific principles in regulating those manufacturing changes are described by internationally accepted guidelines.24 Biosimilar regulation is in fact based on experience with the regulation of manufacturing process changes of originator products.

Figure 1: Variability of major glycan variants in commercially available monoclonal antibody.22
Analytical comparison of molecules sets the foundation for extrapolation of indication for biosimilars. Extrapolation follows the concept that the same molecule will behave the same way as the original molecule in all indications and patient populations. If the totality of the evidence (structural, functional, and PK analyses, and clinical data in at least one sensitive indication) demonstrates that the biosimilar and reference product are highly similar, then extrapolation from one molecule to the other is scientifically justified; i.e. the biosimilar can be safely used in all indications approved for the originator.21,25 Extrapolation to all the originator indications is not based simply on the clinical data alone for a biosimilar in one tested indication, but includes all the analytical data demonstrating that the molecules are structurally and functionally the same.26 Therefore, it can be expected that both will behave the same way in all the indications tested for the originator. It should be noted that extrapolation for biosimilars is not granted automatically but is evaluated for each indication individually based on the totality of evidence.20,21
The concept of extrapolation is not new; it is also applied when the manufacturing processes of originator medicines are changed. For example, if changes in glycation of an originator molecule following manufacturing process changes were identified, the company is required to demonstrate that the changes do not lead to clinically meaningful differences and therefore patients and physicians can expect the same safety and efficacy. If the modified process is approved as producing a highly similar product under the same label, it can be extrapolated to all approved indications.22
Current analytical tools are extremely powerful and able to detect very small variations in molecular structure and function. When demonstrating whether a biosimilar matches the originator in all relevant structural and functional attributes, typically >40 methodologies are used to analyse >100 different attributes.27 Ideally, attributes are measured by more than one method. Using the development of the ETN biosimilar GP2015 as an example, the primary structure was tested by peptide mapping and mass spectrometry, and was shown to be 100% identical in amino acid sequence to ETN samples sourced in Europe and the USA.28 X-ray crystallography confirmed that the higher order structure of GP2015 was indistinguishable from ETN sourced from Europe and the USA. Similar biological activity in neutralising tumour necrosis factor-a (TNF-a) was confirmed in a TNF-a reporter gene assay.29 Structurally and functionally, and considering the batch to batch variability, GP2015 is indistinguishable from ETN and can therefore be expected to have the same clinical activity in all indications approved for the originator.
In conclusion, biosimilarity is established through stringent regulatory licensing pathways based on analytical data and complemented by preclinical and clinical studies confirming structural and functional similarity. Physicians and patients can expect the same clinical efficacy and safety profile for the biosimilar as for its originator. Extrapolation is evaluated for each indication based on the totality of evidence of structural, functional, preclinical, and clinical data. This concept has also been successfully used for originator products following manufacturing process changes. The safety and efficacy of biosimilars has been confirmed by over 10 years’ experience with biosimilar products on the European market.
Demonstrating Clinical Equivalence: Results of the EGALITY Trial
Doctor Sascha Gerdes
In biosimilar development, clinical studies are required to confirm the equivalent efficacy of the biosimilar and its originator. Clinical studies to confirm biosimilarity need a sensitive population, a well-defined primary endpoint, and an adequate study duration to allow detection of small differences in efficacy, safety, and immunogenicity, should there be any. Plaque-type psoriasis fulfils all of these criteria. The response, in terms of reduction in skin lesions, is rapid and easy to assess. PASI and Physicians’ Global Assessment (PGA)-based endpoints are well-established and consistent.30 The treatment effect size compared to placebo is large, and the dosing for ETN in psoriasis is within the linear range of the dose-response curve.6,31 In psoriasis, biologics are usually used as monotherapy, in contrast to indications such as rheumatoid arthritis where co-medication with immunosuppressive drugs is common and drug interactions may occur.32,33
EGALITY is a randomised, double-blind, Phase III confirmatory study conducted in patients with moderate-to-severe psoriasis to compare efficacy, safety, and immunogenicity of GP2015 with ETN. The study also provides data on multiple switches between the originator and the biosimilar, which is relevant for clinical practice.34 The primary endpoint is equivalence of PASI 75 response rates at Week 12 and the main secondary endpoint is equivalence of the mean percent change in PASI score from baseline to Week 12. The study was conducted in 74 dermatology centres in 11 European countries and South Africa. Overall, 531 patients were randomised 1:1 to 50 mg GP2015 or ETN twice weekly (subcutaneous regimen) for 12 weeks (Treatment Period [TP] 1). Patients who reached a PASI 50 response at Week 12 compared with baseline were re-randomised to receive either continuous treatment with ETN or GP2015, or treatment that involved three treatment switches between GP2015 and ETN at 6-week intervals until Week 30 (TP 2), after which they continued on their current treatment until Week 52 (Extension Period) (Figure 2).
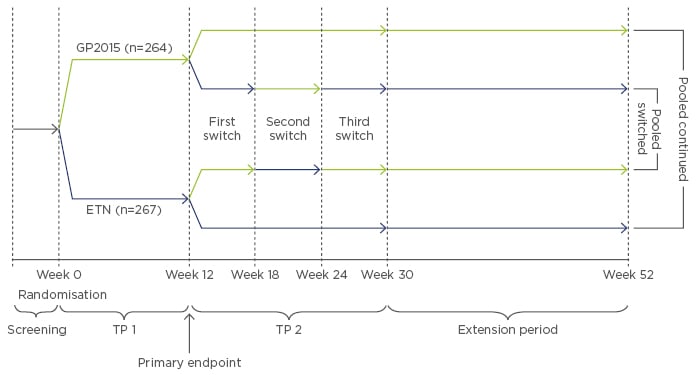
Figure 2: Study design of EGALITY.34
Randomised, double-blind, Phase III confirmatory study of the etanercept biosimilar GP2015 and ETN.
ETN: originator etanercept; GP2015: ETN biosimilar; TP: Treatment Period.
Eligible patients were male or female adults with active but clinically stable chronic plaque-type psoriasis diagnosed at least 6 months prior to enrolment, with ≥10% of the body surface area affected, a PASI score of ³10, and an Investigators’ Global Assessment (IGA) score of ≥3. These patients had to have previously received phototherapy or systemic therapy for psoriasis, or have been candidates for such therapy.34 Patients were excluded if they had other forms of psoriasis, in case of ongoing use of protocol-prohibited psoriasis treatments such as topical corticosteroids or UV therapy, or in case of previous exposure to ETN. Baseline characteristics were similar between the study arms. The mean age was approximately 42 years and BMI was 28.6 kg/m2 in the GP2015 group and 28.5 kg/m2 in the ETN group. The baseline PASI score was quite high, with a mean value of 22.5 in both groups.
PASI 75 (describing a 75% improvement compared with baseline) response rates were equivalent at Week 12, and were achieved by 73.4% of patients receiving GP2015 and 75.7% of patients receiving ETN in the per-protocol set (PPS).34 Overall, PASI response rates (PASI 50, 75, and 90) were highly similar over the first 12 weeks of the trial. The mean percentage change in PASI score from baseline to Week 12 was equivalent between the GP2015 and ETN-treated PPS groups; the least square means difference between the two groups was -0.64% for the mixed-model repeated measures and -0.88% for the averaged treatment effect. The 95% confidence intervals were within the pre-specified margin range (15% to -15%). The PPS was used for the PASI response rates instead of the intent-to-treat or last observation carried forward approach used in pivotal trials testing for superiority, because the PPS is considered the more sensitive population in equivalence or non-inferiority trials. Similar improvements in IGA and Dermatology Life Quality Index (DLQI) scores were achieved in both treatment arms.34
Up to Week 52, PASI response rates were comparable between the groups who continued GP2015 or ETN treatment without switching (Figure 3). There was no impact of treatment switches on PASI response up to Week 52 when pooled data from all patients who underwent repeated switches between GP2015 and ETN were compared with pooled data from all patients who continued treatment without switching. Immunogenicity was low and in line with previously reported rates for ETN; five patients, all in the ETN group, developed anti-drug antibodies (ADAs) in the period up to Week 12, and one further patient who switched from ETN to GP2015 developed ADAs in the extension period up to Week 52. All ADAs were non-neutralising, transient, and low in titre.
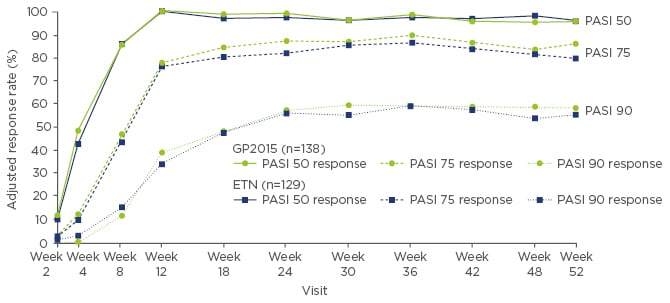
Figure 3: PASI response rates for continued GP2015 and ETN treatment groups from baseline to Week 52 in the overall per-protocol set.34
ETN: originator etanercept; GP2015: ETN biosimilar; PASI: Psoriasis Area and Severity Index.
The safety profiles of GP2015 and ETN were similar over the 52 weeks and were not affected by treatment switching (Table 1). There was no discernible pattern in treatment-emergent adverse events of special interest, such as rash (none with GP2015 versus one with ETN), neoplasms (five with GP2015 versus one with ETN), neutropenia (two with GP2015 versus none with ETN), or infections (eight with GP2015 versus three with ETN).34
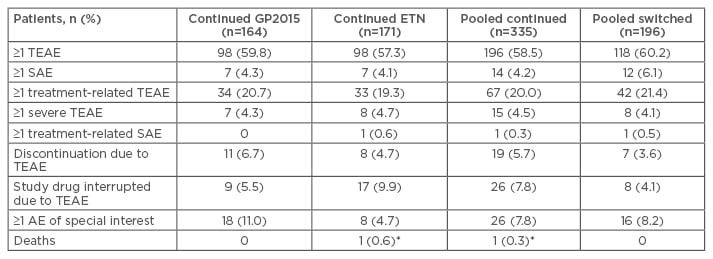
Table 1: Patients with treatment-emergent adverse events in the study groups over the 52-week trial period.34
*Cardiopulmonary failure, considered by investigator to be unrelated to study drug.
TEAE: treatment-emergent adverse events; SAE: severe adverse events; AE: adverse events; GP2015: ETN biosimilar; ETN: originator etanercept.
The EGALITY study confirmed equivalence in the efficacy of GP2015 and ETN in patients with moderate-to-severe, chronic, plaque-type psoriasis by meeting all the primary and secondary endpoints. The safety profiles of GP2015 and ETN were comparable, and no new or unexpected safety issues with GP2015 or ETN were reported. The incidence of ADAs was low. Switching treatments did not impact efficacy, safety, or immunogenicity. These findings provide clinical confirmation of similarity between GP2015 and ETN and contribute to the totality of evidence, confirming that GP2015 is an adequate ETN biosimilar.
Question and Answer Session
Q: Is the triple switch in EGALITY sufficient to get an interchangeability approval on the label in the USA and Europe?
Dr Gerdes replied that the triple-switch study design was unique to this study, and whereas a single switch in a study was probably not sufficient to address whether switching affects safety or efficacy, the triple switch in EGALITY had clearly shown that it did not impact efficacy.
Q: Do the manufacturers of originator biologics have more scope for improvements in functionality resulting from manufacturing process changes than manufacturers of biosimilars?
Dr Schiestl replied that for the originator, the goal was still to keep the same safety and efficacy profile. Regulatory agencies will only approve a manufacturing process if the safety and efficacy stay the same. If the manufacturing process changes the molecule such that clinically meaningful differences occur, then the company has to do additional trials for safety and efficacy, and the molecule will be approved with a different name and label with its own safety data.





