
Meeting Summary
Prof Costanzo welcomed attendees to the symposium and outlined the agenda before explaining the autoimmune pathophysiology that underlies psoriasis. Dr Caprioli discussed the role of the T helper (Th)17 cell lineage and its accompanying cytokines in autoimmune diseases. Prof Paul discussed the latest clinical trial data for therapies targeting the interleukin (IL)-17/IL-23 axis in patients with psoriasis, and how this is changing the treatment landscape. The symposium was followed by a question and answer session.
The Autoimmune Story of Psoriasis: A Focus on Autoantigens and Cytokines
Professor Antonio Costanzo
Psoriasis is a multifactorial autoimmune disease with various genetic and environmental stimuli contributing to its pathogenesis.1 Therefore, identifying genetic markers, functional networks, and regulatory mechanisms that drive disease progression may aid the development of novel therapeutics.
Genome-wide association studies have identified significant associations between 63 genetic loci and psoriasis, accounting for approximately 28% of the estimated disease heritability.2 Most of the genes identified in these studies are related to the immune system, particularly the regulation of lymphocyte differentiation, responses to viruses and bacteria, signalling, and pattern recognition.2 For example, variants of the HLA-Cw6 gene, which codes for a component of the major histocompatibility complex, have been implicated in early onset (Type I) psoriasis because antigen-presenting cells that express HLA-Cw6 variants are known to recognise and bind both the streptococcal M protein and keratin found in the skin.3 Consequently, HLA-Cw6 allele expression can result in an autoimmune response to fragments of keratin protein, activating both the innate and adaptive immune responses.3 Furthermore, several other immune-related genes associated with psoriasis (e.g. the gene coding for the IL-23 receptor) have been linked to chronic autoimmune and inflammatory disorders, including Crohn’s disease and ankylosing spondylitis.3
Keratinocytes are also believed to play a central role in psoriasis because changes in the expression of genes related to maintaining the epidermal barrier in these cells (e.g. cJun and JunB) have also been implicated in the pathology of psoriatic lesions.4-6 Activated keratinocytes and other innate immune cells release cytokines that activate myeloid dendritic cells, generating innate and adaptive immune responses.7 In addition, keratinocytes produce a range of antimicrobial peptides (AMP), such as LL-37 (a 37-amino acid cathelicidin AMP) and β-defensins, as the first line of defence against microbial infiltration through the skin.6,7 Notably, AMP are highly expressed in psoriatic skin, activate proinflammatory pathways, and shape the immune microenvironment.6,8
LL-37 forms complexes with DNA released from injured cells, encouraging the formation of condensed DNA–LL-37 aggregates; however, in up to 75% of patients with moderate-to-severe psoriasis, circulating CD4+ and CD8+ T cells recognise LL-37 as an autoantigen.8,9 Keratinocytes and/or epidermal plasmacytoid dendritic cells (pDC) possessing an HLA-Cw6 allele may present LL-37 to autoreactive CD8+ T cells (e.g. Th1/Th17 cells and natural killer cells), which in turn release inflammatory cytokines, such as interferon (IFN)-γ and IL-17, activating the adaptive immune system.9 In addition, mature pDC secrete IL-12 and IL-23, which induce naïve T cells to differentiate into Th1 and Th17 cells.7,9
IL-26, produced by Th17 cells, has direct bactericidal properties and can form insoluble complexes with self and bacterial-DNA released from dying cells;10 IL-26–DNA complexes are thought to activate more Type I IFN-producing pDC in a similar manner to LL-37.10 Consequently, self-DNA-bound IL-26 may contribute to a perpetual inflammatory feedback loop, resulting in autoimmunity.
CD8+ T cells isolated from psoriasis plaques also recognise a melanocyte-expressed molecule known as ADAMTSL5 when it is presented on the HLA-Cw6 protein, suggesting the existence of a second autoantigen.11 Indeed, both ADAMTSL5 and LL-37 are highly expressed in cases of active psoriasis and often co-expressed within dendritic cells and keratinocytes.12 Therefore, given the central role of LL-37 in the autoimmune response observed in patients with psoriasis, LL-37 offers a potential treatment target.
In conclusion, existing studies have demonstrated that psoriasis is an autoimmune disorder that results in inflammatory cytokine-induced skin and systemic damage.1,3 While promising susceptibility markers for early onset psoriasis (e.g. HLA-Cw6) and autoantigens that stimulate immune cell activation (e.g. LL-37 and ADAMTSL5) have been identified,3,7-9,11 further research is required to investigate further into the complex biochemical networks that drive autoimmunity in patients with psoriasis. Understanding the biochemical networks that underlie psoriasis may help deliver additional therapeutic targets and novel treatment options.
Lessons from Interleukin-23 in Immune-Mediated Inflammatory Diseases
Doctor Flavio Caprioli
According to the classical Th1/Th2 immune response paradigm, naïve T cells differentiate into Th1 or Th2 lineages, each with a unique cytokine secretion profile driving cell-mediated immune responses or antibody-mediated responses, respectively.13,14 Therefore, human autoimmune diseases have generally been separated into two classes based on experimental models: Th1-predominant diseases, such as psoriasis and Crohn’s disease, or Th2-predominant diseases, such as allergic asthma and ulcerative colitis.13 Similarly, experimental models of autoimmune diseases have been classically divided into Th1 or Th2-mediated diseases. For example, administering myelin antigens and Freund’s complete adjuvant to otherwise healthy mice results in experimental autoimmune encephalomyelitis (EAE), a severe and potentially fatal neurological disease resembling human multiple sclerosis.15 In this model, the central nervous system is heavily infiltrated with the IFN-γ-producing CD4+ T cells, leading to the initial hypothesis that EAE was a Th1-mediated disease. However, subsequent work with IFN-γ knockout mice resulted in a worsening of EAE, as opposed to the anticipated disease-resistance, suggesting a need for a shift in our understanding of the Th1/Th2 paradigm.16
As IL-12 is an important factor in the formation of Th1 cells, it was initially considered to be an essential factor in the EAE model.17 However, following the discovery and cloning of IL-23, which is comprised of a p19 subunit and a p40 subunit that is shared with IL-12, it was instead determined that IL-23 was the critical factor driving this model.17 Studies using IL-10-deficient mice confirmed that the presence of p19, but not p40, was essential in maintaining a chronic inflammatory state.18 It was later elucidated that IL-23 was a critical factor to confer pathogenic features to Th17 cells, which differentiate from naïve CD4+ T cells under the combined action of transforming growth factor-β and IL-6.19,20 Under homeostatic conditions, IL-23 is mainly expressed in the bone marrow and gastrointestinal tract (Figure 1).21 Conversely, an overexpression of p19 subunit is detected in the gastrointestinal tract in patients with active inflammatory bowel disease (IBD) and in the skin of patients with active psoriasis.22,23 The reasons behind IL-23 overexpression in these clinical conditions are still unclear, in spite of recent hypotheses about changes in the intestinal microbiota. Moreover, mutations in the IL-23 receptor increase susceptibility to several human autoimmune diseases, suggesting that a link between autoimmune inflammation and increased biological responsiveness to this cytokine exists.24
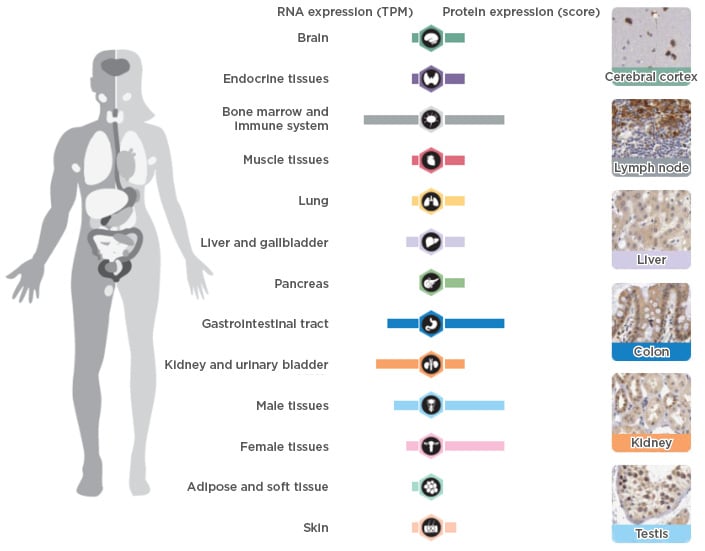
Figure 1: RNA and DNA expression of interleukin-23 in healthy humans.
TPM: transcripts per million.
Adapted from The Human Protein Atlas21
Cytokines secreted by Th17 cells, particularly IL-17, tumour necrosis factor (TNF), and IL-22, can have pleiotropic pathogenic effects in psoriasis and other autoimmune diseases. Specifically, IL-17 induces psoriasis-related gene transcription and activates inflammatory cells, encouraging neutrophils to infiltrate the skin, resulting in inflammation and reactive proliferation of both keratinocytes and blood vessels and ultimately leading to psoriasis plaque formation.25 IL-17 is also responsible for mediating inflammation, enthesitis, and bone erosion in the joints of patients with psoriatic arthritis.26-29 However, targeting IL-17 expression may be difficult because the cytokine is required for tight junction integrity in the intestinal epithelium and this may explain why anti-IL-17 treatments have not yet proven to be effective for IBD.30
In summary, the discovery of Th17 cells, IL-17, and IL-23 has changed our understanding of the underlying pathology of autoimmune diseases, such as psoriasis and IBD. Experimental data and ongoing clinical studies have highlighted the potential of the IL-17/IL-23 axis as a therapeutic target for treating patients with psoriasis and psoriatic arthritis.
Psoriasis: Linking Pathophysiology and Clinical Evidence
Professor Carle Paul
Until 1980, psoriasis was thought to be a keratinocyte hyperproliferative disease, but between 1980 and 1990, a link between psoriasis, cytokine expression, and keratinocyte proliferation was identified.31,32 In particular, psoriasis was classified as a Th1-mediated disease because cyclosporine treatment was effective in some cases. It was not until 1994, when elevated TNF-α levels in psoriatic lesions were reported, that the first proof-of-concept study targeting cytokines could be performed.31,32 This was followed by the identification of IL-12 and IL-23 in the decade from 2000–2010, and the Th17 cell lineage, which also facilitated a proof-of-concept study supporting targeting of the Th17 pathway in patients with psoriasis. Notably, over the last 12 years our understanding of the underlying pathophysiology of psoriasis has evolved, from psoriasis being the result of injury and then the activity of IFN-γ and Th1-mediated inflammation,33 to the current model that considers the roles of dendritic cells in producing cytokines and multiple effector cells, particularly Th17 cells.34
Likewise, ongoing research is likely to further elucidate the pathological pathways that contribute to psoriasis, resulting in a rapid rate of treatment innovation. For example, traditional immunosuppressants were the only available therapy in the 20th century, whereas between 2000 and 2010, targeted anti-TNF-α therapies were developed. In addition, since 2010, five new biological therapies have been approved for the treatment of psoriasis in the USA, and an even wider range in Europe, while many other novel therapies continue to progress through Phase II and III clinical studies.
There are currently no clear biomarkers to guide treatment selection for patients with psoriasis, but clinicians tend to have a multidimensional approach to treatment, considering the efficacy and safety profile of each agent, patient comorbidities, and patient preference. For example, each of the anti-IL-17 agents (secukinumab, ixekizumab, and brodalumab) is generally efficacious, with the relative efficacy dictated by pharmacokinetics, i.e. differences in exposure to each drug. However, while up to 40% of patients will experience complete clearance of psoriasis, relapse will often occur soon after ceasing treatment.35-38
One anti-IL-23 therapy (guselkumab) has been granted marketing approval for the treatment of psoriasis in the USA, while tildrakizumab and risankizumab are under development. Administering guselkumab 100 mg at Weeks 0 and 4, followed by dosing every 8 weeks, resulted in almost 75% of patients achieving a 90% reduction in Psoriasis Activity Severity Index (PASI 90) compared with <3% of patients treated with placebo, and 85% of patients having an Investigator’s Global Assessment (IGA) of 0 or 1 versus 7% for placebo (Figure 2).39 In addition, the response was maintained for ≥2 years.39 A greater proportion of patients also responded to guselkumab compared with adalimumab and no loss of response over time was seen with guselkumab (Table 1). Response rates are also increased by switching patients who have not achieved a full response after 16 weeks of ustekinumab treatment to guselkumab.40 Complete clearance (PASI 100) at 2 years was also observed in approximately half of all patients treated with guselkumab and responses were maintained for >6 months after ceasing therapy in a relatively high proportion of patients.41 Furthermore, the high level of response was achieved with guselkumab therapy without increasing the risk of infection, particularly opportunistic infections, such as Candida and Staphylococcus aureus.39
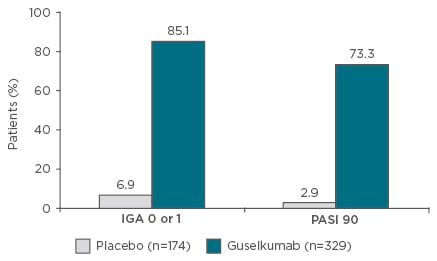
Figure 2: VOYAGE I Phase III results.39
Guselkumab is not approved for the treatment of psoriasis or psoriatic arthritis in Europe. Co-primary endpoints: IGA 0 or 1 and PASI 90 at Week 16. Eligible patients had moderate to severe plaque psoriasis (PASI ≥12; PGA ≥3; body surface area ≥10%). p<0.001.
IGA: Investigator’s Global Assessment; PASI: Psoriasis Area Severity Index; PGA: Physician’s Global Assessment.
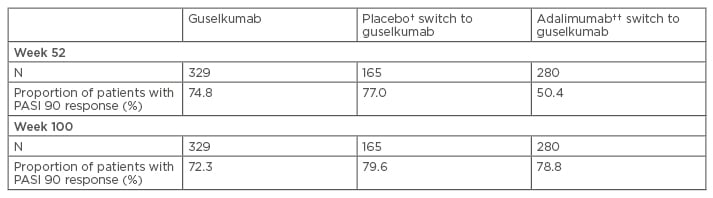
Table 1: Proportion of patients who achieved Psoriasis Area Severity Index 90 response (Week 0–100).*
*Data was inputed for non-responders; †Patients were treated with placebo and switched to guselkumab at Week 16; ††Patients treated with adalimumab for the first 48 weeks then switched to guselkumab.
PASI: Psoriasis Area Severity Index.
Risankizumab 180 mg has demonstrated similar results to guselkumab in Phase II studies, with 81% of patients achieving PASI 90, while Phase III studies are ongoing.42 In contrast, approximately 60% of patients treated with tildrakizumab, an anti-IL-12/IL-23 antibody, achieved PASI 90 or IGA 0 or 1 in a Phase III study, but the response rates increased slightly after long-term follow-up.43 In addition, following clinical research outcomes in other autoimmune diseases, such as IBD, a hypothesis paper has been developed suggesting that early and aggressive intervention for patients with psoriasis could modify the course of the disease and improve patient outcomes.44 Anti-IL-23 therapies are suitable candidates for testing this hypothesis. In conclusion, anti-IL-23 therapy may offer similar efficacy to anti-IL-17 therapy, with the potential benefit of a sustained response after ceasing treatment. This not only provides the opportunity to increase dosing intervals for some patients, but to also investigate the disease-modifying potential of anti-IL-23 therapy in future studies.
Question and Answer Session
Q: If psoriasis is classified as an autoimmune disease, why is the condition observed in limited areas of the skin and not in all areas?
A: Dr Caprioli noted that this is an unresolved question. One explanation could be trauma as a trigger for psoriasis; differences in microbiota across the skin could be another explanation.
Q: Is there still a role for topical treatments, such as corticosteroids and vitamin B preparations, in treating patients with psoriasis?
A: Dr Caprioli suggested that topical treatments are appropriate for patients with mild psoriasis because there is no systemic involvement in these cases.
Q: Could combination therapy with two biological therapies be effective in treating patients with psoriasis?
A: Dr Caprioli outlined how biological combination therapies are being investigated in patients with highly refractory IBD, with a particular focus on patients with comorbidities. The efficacy of these treatment options in autoimmune disease has not yet been established, but the potential immunosuppression-related adverse events could be a barrier to routine use of biological combination therapy.
Q: Is there a neurological component to psoriasis?
A: Prof Costanzo highlighted the potential link between stress and psoriasis, while Dr Caprioli indicated that, in his opinion, there may be a neurological element to psoriasis which needs to be explored further.
Q: What is the biggest challenge for the treatment of patients with psoriasis in the future?
A: Prof Paul indicated that the goal is to discover treatments that are long-acting and/or disease-modifying so that the severity of the disease experienced by some patients can be permanently controlled.
Q: Are there any biomarkers that can be used to guide the treatment of psoriasis with targeted therapies?
A: Prof Costanzo replied that biomarkers are yet to be identified, but this is an area of active research.
Click here to view the full symposium.





