
Meeting Summary
The satellite symposium comprised two short presentations aimed at providing an overview of the rationale for the use of interleukin (IL)-23 inhibition as a targeted strategy to treat immune-mediated inflammatory diseases. Presentations by Prof Prinz and Prof Danese focussed on psoriasis and inflammatory bowel disease, respectively, as examples of clinical indications in which the gene-to-clinic approach has led to the development and approval of biologic IL-23 inhibitors. In psoriasis the introduction of targeted anti-IL-17/IL-17 receptor A-chain (RA) and anti-IL-23 biologic therapies has provided a paradigm shift in the management of the disease, making complete clearance of disease a realistic aim for the first time. The use of IL-12/IL-23 inhibitors, such as ustekinumab, is now also possible in Crohn’s disease (CD), providing another example of the successful translation of immunological targeting into clinical practice.
Interleukin-23 Inhibition as a Strategy to Treat Immune-Mediated Inflammatory Diseases: A Focus on Psoriasis
Professor Jörg Christoph Prinz
Effector T cells have evolved into different functional subsets, each with distinct physiological roles and signature cytokine profiles.1-3 T helper 17 (Th17) cells are a functional lymphocyte subset that has developed to co-ordinate the immune response against bacterial and fungal infections and are characterised by the production of IL-17, IL-22, and interferon (IFN)-γ. As well as providing a key protective role in host immunity, Th17 can also have a pathogenic role in various autoimmune diseases, including systemic lupus erythematosus, multiple sclerosis, rheumatoid arthritis, psoriasis, and inflammatory bowel disease (IBD).3 Specific and targeted inhibition of Th17-mediated immune pathways has therefore emerged as a highly effective treatment approach for psoriasis and IBD, with a number of biologic agents being developed and licensed for these indications.
Differentiation of Th17 cells from naïve cluster of differentiation 4-positive (CD4+) T cells occurs in three distinct stages.4 Upon activation of T cells, transforming growth factor-β and IL-6 establish early commitment to the Th17 lineage by activating signal transducer and activator of transcription 3 (STAT3), which induces the expression of IL-21. Autocrine signalling by IL-21 then promotes STAT3-dependent expression of the master transcription factor for Th17 differentiation, retinoic acid receptor-related orphan receptor gamma t (RORγt), leading to the production and expression of IL-17A and the IL-23 receptor. This allows IL-23 to bind to and exert its effects on previously committed Th17 cells, stabilising the phenotype and expansion of Th17 cells, which secrete the effector cytokines IL-17A, IL-17F, IL-21, IL-22, and IL-26. The development of biologic agents that target the Th17 pathways has used several approaches. One approach is to inhibit the Th17 effector response, either by inhibiting the production of IL-17A/IL-17F using anti-IL-17 antibodies or by inhibiting the signalling of IL-17A/F through the blockade of the IL-17 receptor alpha chain. More recently, an alternative strategy has emerged: the inhibition of IL-23 using anti-IL-23 antibodies to interfere with the stabilisation and expansion of Th17 cells.
The IL-17 family consists of six cytokines (IL-17A–F), which signal through a family of heterodimeric IL-17 receptor complexes, i.e., receptors composed of two different chains. Receptors for IL-17A, IL-17F, IL-17C, and IL-17E are composed of the IL-17RA chain and one of three different other chains to form a functional receptor unit. The IL-17RA chain combines with the IL-17-RC chain for binding of IL-17A and IL-17F, with the IL-17RE chain for binding of IL-17C, and with the IL-17RB chain for binding of IL-17E (Figure 1).6 Antibodies directed against IL-17A selectively neutralise IL-17A and the IL-17A/F heterodimer.5,7,8 However, as the IL-17RA chain is part of several heterodimeric IL-17 chain receptors, blocking IL-17RA interferes with the signalling of most members of the IL-17 cytokine family, including IL-17C and IL-17E, and therefore IL-17RA blocking has a much broader effect. This broader inhibitory activity may explain the greater efficacy that has been observed with antibodies directed against the IL-17RA chain compared with those against IL-17A. This improvement in efficacy is exemplified by the comparative efficacy of brodalumab (an anti-IL-17RA antibody) and secukinumab (an anti-IL-17A antibody) in the treatment of chronic plaque psoriasis. Although no head-to-head clinical trials have been conducted, Phase III clinical data from the AMAGINE-2 and AMAGINE-3 (both brodalumab) and ERASURE (secukinumab) studies show that both treatments are highly effective in patients with plaque psoriasis, with approximately 80% of brodalumab-treated patients and 65% of secukinumab-treated patients becoming clear or almost clear of psoriasis.9,10 However, a greater proportion of patients treated with brodalumab attained ≥75% improvement in Psoriasis Area Severity Index (PASI) 75, PASI 90, and PASI 100 responses as compared with secukinumab.9,10 Importantly, the response can be maintained over time, which has been shown in recently published data showing that psoriasis treatment with secukinumab is associated with sustained PASI responses through 3 years of treatment.11
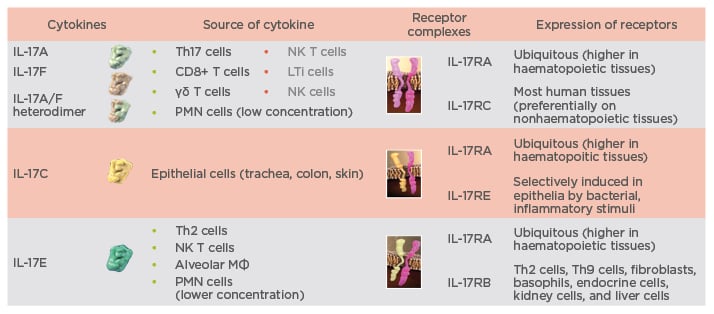
Figure 1: Spectrum of targets for interleukin-17A and interleukin-17RA antibodies.5-8
IL: interleukin; LTi: lymphoid tissue inducer; MФ: macrophages; NK: natural killer; PMN: polymorphonuclear.
leukocyte; R: receptor; Th: T helper.
An alternative approach to blocking the Th17/IL-17 effector response is to block the upstream cytokine IL-23. As mentioned previously, inhibition of IL-23 interferes with the stabilisation and expansion of Th17 cells without affecting the differentiation of Th17 populations and consequently IL-23 inhibition has regulatory effects on memory effector T cells (i.e., those involved in the pathogenic response), but not on naïve or central memory T cells.4 Currently, clinical data are available for three IL-23 inhibitors in the treatment of psoriasis: tildrakizumab, guselkumab, and risankizumab. Although differences in the overall response to each of the IL-23 antibodies have been observed in clinical trials, blocking IL-23 appears to be highly efficacious. An improvement of >90% in PASI is realistic and clinical trials have reported that, at Week 12 or 16 (depending on the study), 12−14% of patients treated with tildrakizumab, 34−37% of patients treated with guselkumab, and 48% of patients treated with risankizumab achieved complete clearance of psoriasis (i.e., PASI 100 response).12,13 Tildrakizumab seems to be associated with a slower initial clinical response, but the efficacy appears to ‘catch-up’ with the other IL-23 inhibitors over time, and 67–69% of patients achieve a Physicians’ Global Assessment of 0 or 1 (indicating clear or almost clear of psoriasis) by 28 weeks.12,13 However, it is important to note that these data are not from head-to-head comparisons of the IL-23 inhibitors and the outcomes reported for risankizumab are from a Phase II clinical trial. Clinically, the effect of blocking IL-23 has been shown to be superior to blocking tumour necrosis factor (TNF)-α.14 The VOYAGE 1 trial, which compared guselkumab with the anti-TNF-α inhibitor adalimumab in patients with moderate-to-severe psoriasis, reported that specific interference with the maintenance of activation of Th17 cells via IL-23 inhibition achieved PASI 75, PASI 90, and PASI 100 responses in a significantly higher percentage of patients than did TNF-α blockade.14
Although blocking either IL-17 or IL-23 targets the same Th17 effector pathway, there is a difference in the effects of each of these approaches on the immune response.15-18 Blocking IL-23 has been shown to be very effective in both psoriasis and CD, whereas anti-IL-17A or IL-17RA antibodies are highly effective for the treatment of psoriasis but may exacerbate CD in a subset of patients. This effect has been reproduced in a mouse model of colitis, in which IL-17 inhibition weakened intestinal epithelial barrier function and increased inflammation, while IL-23 inhibition enhanced regulatory T cell accumulation and attenuated inflammation. It is therefore important to be aware of this possibility because psoriasis and IBD are associated and can develop concurrently in some patients.
Comparison of the dosing regimens of the targeted biologic agents used in psoriasis highlights another interesting issue. Blocking the effector cytokines TNF-α and IL-17 requires more frequent and potentially higher doses of inhibitory antibodies than upstream interference with the regulation of Th17 activation through the inhibition of IL-23. Antibodies against TNF-α (adalimumab), IL-17 (secukinumab), and IL-17RA (brodalumab) have dose intervals of 2 or 4 weeks, while for anti-IL-23 antibodies (guselkumab, risankizumab) dosing intervals of up to 12 weeks are sufficient to maintain clinical response.13,19-23 Adalimumab and guselkumab have similar serum half-lives (approximately 10–20 days);19,23 however, achieving a sufficient response with adalimumab requires much more frequent dosing than with guselkumab. A 5 mg dose of guselkumab given four times over 40 weeks was sufficient to achieve a Physicians’ Global Assessment of 0 or 1 in up to 40% of patients.24,25 The differences are even more intriguing when the pharmacokinetics of guselkumab are examined. Although the mean serum concentration of guselkumab is almost zero 50 days after a 5 mg dose, a treatment response is maintained;25,26 in other words, clinical efficacy outlasts the presence of the biologic inhibitor. From an immunological perspective, these data indicate that blocking effector cytokines, such as TNF-α, is associated with a different mechanism of action than the inhibition of IL-23; IL-23 antibodies appear to downregulate ongoing Th17 responses and provide disease control beyond the actual presence of active substance. This may be the major difference between blocking effector cytokines and blocking IL-23.
In summary, the extremely high clinical efficacy of IL-23 and IL-17 pathway inhibition has set a new standard for the treatment of plaque psoriasis and, for the first time, achieving complete clearance of disease has become a realistic treatment goal for many patients. Importantly, blocking IL-23 interferes with the maintained activation of Th17 cells and Th17 differentiation, and preferentially regulates the memory effector cells involved in the pathogenic immune response. This is a different therapeutic pathway to inhibition of IL-17 or IL-17RA and long-term follow-up of the clinical effects of sustained IL-23 inhibition is required to assess potential safety benefits of IL-23 inhibition compared with direct inhibition of the IL-17 effector response.
Interleukin-23 Inhibition as a Strategy to Treat Immune-Mediated Inflammatory Diseases: Evidence from the Treatment of Inflammatory Bowel Disease
Professor Silvio Danese
Although the exact cause of IBD is not entirely understood, it is believed to involve a complex interaction between genes, the immune system, and environmental factors. Genetic susceptibility, the composition of the gut microbiome and an inappropriate immune response can all play a role in the development of IBD.27 Indeed, genome-wide association studies have revealed major genetic variations in the IL-23 receptor and the IL-12 p40 subunit, both of which are involved in the immune inflammatory response in patients with CD and ulcerative colitis (UC). However, although there is a strong genetic susceptibility for IBD and >163 genetic associations for IBD have been identified, these account for <30% of all cases of IBD.27 An inappropriate immune response is responsible for the development of IBD in the majority of patients and a targeted inhibition of key immune-mediated inflammatory pathways has emerged as a leading new therapeutic strategy. Among the plethora of potential lymphocyte and effector cytokine targets, T cells are the key drivers of the pathophysiology of IBD from early to late disease.28 Although the immune pathways associated with IBD have many similarities with other autoimmune inflammatory diseases, such as psoriasis, there are patterns of cytokine-mediated pathology that are specific to IBD. Firstly, it must be noted that inhibition of effector cytokines in the Th17 pathway (e.g., with anti-IL-17A antibodies) can exacerbate inflammation in some patients with CD, as Prof Prinz explained previously, and this is important to remember when using such biologics in the clinic. With regard to the immunopathology of IBD, there is clinical evidence that shows upregulation of IL-12 occurs in patients with early CD compared with those who have UC or healthy controls.29,30 Upregulation of IL-23 then occurs once CD is established.30,31 A study in mice has shown that IL-12 continues to contribute to chronic intestinal inflammation during established colitis and consequently IL-12 has been identified as a key therapeutic target.32 Additionally, experimental colitis models have shown that treatment with anti-IL-23 antibodies attenuated extensive inflammation in both the caecum and colon, and reduced inflammatory infiltrates and epithelial hyperplasia.33 IL-23 inhibition has therefore also been identified as a key target for targeted biologic treatment in CD.
A number of biologic treatments are being developed for targeted inhibition of IL-12 and IL-23 in patients with CD (Figure 2). Ustekinumab, the first such biologic to be approved for the treatment of CD, targets the p40 subunit that is present on both IL-12 and IL-23. Risankizumab is another biologic that is in clinical development and targets the p19 subunit, which is present on IL-23 but not on IL-12 (Figure 2).
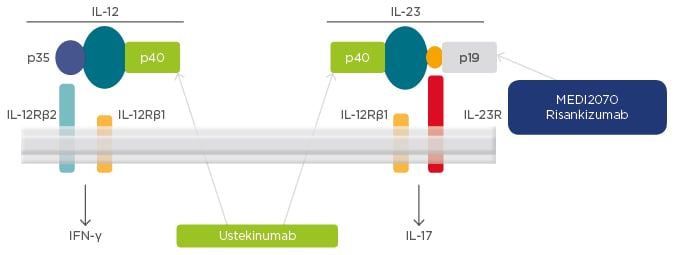
Figure 2: Targets for interleukin-12 and interleukin-23 inhibition in Crohn’s disease.
IFN: interferon; IL: interleukin; R: receptor.
Phase III clinical data have shown that the approved dose of ustekinumab, 6 mg/kg, was associated with a clinical response in a significantly higher proportion of patients than placebo in both patients who had previously failed treatment with anti-TNF-α and those who had previously failed conventional treatment (Table 1).34 Maintenance treatment with ustekinumab has also been shown to be effective in maintaining clinical remission in CD, administered as a subcutaneous dose of 90 mg either every 12 weeks or every 8 weeks.35 Similar clinical data are emerging for risankizumab, which is currently in clinical development and awaiting approval for the treatment of CD.
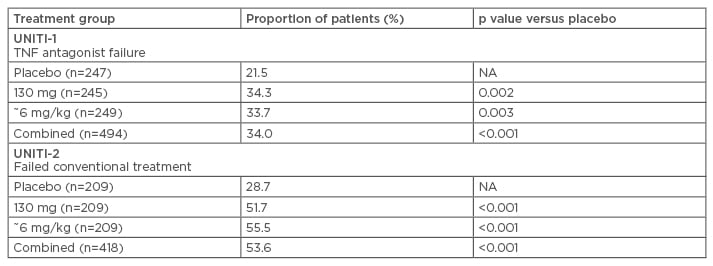
Table 1: Response to ustekinumab in patients with Crohn’s disease who have previously failed anti-tumour necrosis factor-α or conventional treatment (Week 6).34,35
NA: not applicable; TNF: tumour necrosis factor.
To conclude, the development of IL-12 and IL-23 targeted inhibitors is a classic example of the gene-to-clinic approach, providing an effective, novel therapeutic strategy in CD. Clinical studies are ongoing to evaluate the efficacy of IL-12 and IL-23 inhibitors for the treatment of UC, and the publication of the clinical data is eagerly awaited.
Question and Answer Session
Q: In dermatology, when you clear a lesion with a systemic drug there is usually one lesion that stands out and recurs, and that recurrent lesion is frequently the very first lesion that the patient had. This could be a residual lesion that is somehow different to the rest of the skin lesions; do you see that in UC as well, or in IBD?
A: Prof Danese replied that this is a great point and is also seen in patients with CD. For example, when a patient with CD is given an anti-TNF-α drug, healing is observed in the ileum but not in the rectum, and treatment with two drugs is needed because the disease is driven by different mechanisms of action in the different sites. Currently, we have little understanding of zonal gene expression and the mechanisms that drive inflammation in the gut, and this should be the focus of research efforts to understand the differences between different disease sites and locations in order to determine effective drug treatment combinations. Prof Prinz also commented that in dermatologic indications there appears to be a residual scar or residual lesion that is characterised by a greater tendency to restart inflammation, potentially due to low levels of residual inflammation.
Q: As alluded to in the symposium, there are patients who have CD and develop paradoxical psoriasis, and patients with psoriasis who develop CD. Do you have any insights into the genetics, the immunology, and the management of those patients?
A: Prof Danese replied that, at present, although the clinical characteristics of these patients have now been elucidated, as yet nothing is known about the underlying genetics in such cases. We only know that the disease is somehow driven, again, by IL-23 and that these patients respond very well to ustekinumab treatment.
Click here to view the full symposium.





