
Meeting Summary
This symposium, which took place during the 2018 meeting of the European Academy of Dermatology and Venereology (EADV) in Paris, France, provided an overview of the IL-23 revolution in psoriasis, with a specific focus on psoriasis pathogenesis and its relation to potential treatment targets and the development of novel targeted immune therapies. The session focussed on the discovery and development of IL-12 and IL-23-targeted therapies for psoriasis, the role of IL-23 in disease control, and the implications of recent data for clinical practice.
An increasing number of potential treatment options are becoming available for psoriasis, and the differential effect of these agents on various signalling pathways has facilitated a greater understanding of the molecular mechanisms driving disease progression. The symposium initially explored the central role of IL-23 in psoriasis, the mode of action of the monoclonal antibody (mAb) guselkumab in targeting this heterodimeric cytokine, and the parameters associated with a maintenance of response in patients with psoriasis undergoing treatment. The speakers subsequently reviewed current data relevant to the blockade of IL-23 versus dual blockade of IL-12/23, or blockade of the downstream effector IL-17, and the relative effects of these different strategies in psoriasis at the molecular and cellular levels. The concept of ‘disease memory’ in psoriasis was also explored, with an examination of recent data of patients with long-lasting remission, and disease models and future investigations discussed.
Introduction
Dermatologists need to understand the unmet needs in the management of psoriasis and how current data from recently approved or pipeline compounds can help address these needs in clinical practice. This symposium aimed to promote an understanding of psoriasis pathogenesis and its relation to the development of novel targeted immune therapies. The presenters discussed how treatment strategies could be used to optimise long-term patient outcomes and addressed the concept of potential disease modification effects of targeted therapies in psoriasis.
The Road of Discovery: IL-12 and IL-23-Targeted Therapies in the Treatment of Immune-Mediated Inflammatory Diseases
Doctor Ernesto Muñoz-Elías
The proposed model for the immunopathology of psoriasis was, until recently, based on an equal contribution of IL-12 and IL-23 when produced by activated macrophages and dendritic cells. In this model, IL-12 activates Th1 cells and IL-23 activates both Th17 and Th22 cells, which leads to the proliferation of keratinocytes, production of multiple proinflammatory cytokines, increased inflammation, and the formation of psoriatic plaques. However, accumulating data from various sources suggest that the most important driver of pathogenesis in psoriasis is IL-23 rather than IL-12.1 For example, gene expression data show psoriasis lesions have raised expression levels of genes encoding IL-23 (p19, a unique subunit of IL-23, and p40, a subunit of both IL-23 and IL-12) compared with a gene encoding a subunit associated with IL-12 only (the p35 subunit).2 In addition, clinical data showed that the blockade of IFN-γ (primarily a downstream cytokine of IL-12) with anti-IFN-γ was not efficacious in treating psoriasis.3,4 Furthermore, in a knockout mouse model in which IL-12 was silenced, IL-12 was shown to have a protective role in psoriasis-like disease.5 Molecular data show that the first-in-class mAb guselkumab, which binds specifically to the p19 subunit of IL-23, blocks IL-23 signalling while having no effect on IL-12 signalling.6 The downstream production of IL-17 by IL-17-expressing CD8+ T (Tc17) cells, when blocked by a mAb with specificity for IL-17A, such as secukinumab or ixekizumab, precludes the keratinocyte activation that is characteristic of psoriasis.7 Ongoing studies are evaluating the possible effects of IL-23 in multiple immune cell types.
Data from clinical studies are being evaluated to gain insights into the effect of guselkumab on cytokines downstream of IL-23. Response to guselkumab has been examined in patients with moderate-to-severe psoriasis in the Phase III VOYAGE 1 and 2 trials. In the VOYAGE 1 study8 (N=837), patients receiving guselkumab achieved a Psoriasis Area Severity Index (PASI) 90 response rate of 76.3% after 48 weeks of treatment, with superior response rates to adalimumab (47.9%; p<0.001). Guselkumab significantly reduced the levels of key serum effector cytokines, including IL-17A, IL-17F, and IL-22, in the IL-23 pathway at 48 weeks compared with adalimumab.9 The psoriasis transcriptome of patients from VOYAGE 1 was also analysed. Following treatment with guselkumab, an improvement was observed at 4 weeks, 24 weeks, and 48 weeks, and at the 24 and 48-week timepoints, the profile resembled that of non-lesional skin.9 Improvement of the psoriasis transcriptome was more prominent in patients treated with guselkumab than adalimumab. When evaluating multiple gene sets relevant to inflammation, similar results were observed.10 One limitation of whole skin biopsy gene expression analysis is that it does not allow for the characterisation of a drug’s effect on immune cell numbers or phenotypes. Therefore, methods have been developed that allow the dissociation of skin biopsies into single cell suspensions that can then be analysed by flow cytometry for surface and intracellular protein expression. Skin-resident T cells isolated from biopsy samples have been examined, showing that epidermal T memory cells are pathogenic producers of IL-17A, IL-17F, TNF-α, and IL-22.11 Fluorescence-activated cell sorting analysis of skin immune cells represents a new approach for understanding drug effects on skin tissue immune cells and is being incorporated into ongoing studies.
Maintenance of clinical response (PASI 90) after withdrawal of guselkumab has been evaluated in the VOYAGE 2 study,12 in which patients who had received 20 weeks of guselkumab treatment and achieved PASI 90 at 28 weeks were randomised to receive continued guselkumab or switch to placebo. PASI 90 response rates at Week 48 were significantly greater in those receiving continued guselkumab therapy versus those who were withdrawn from therapy (p<0.001); however, 36.8% of patients in the withdrawal arm maintained a PASI 90 response at Week 48 (28 weeks after the last guselkumab dose). Compared with maintained response, loss of response (PASI <75) among patients in the withdrawal arm was associated with significantly increased levels of serum IL-17A, IL-17F, and IL-22 at Week 48.13 Conversely, parameters associated with maintenance of PASI 90 following guselkumab withdrawal included a shorter duration of disease, lower BMI, and lower IL-17F at baseline, as well as complete skin clearance and higher guselkumab concentration at Week 28.14 Further models of single and combined parameters and biomarkers are being investigated to better understand response to guselkumab and the mechanisms behind its action.
In conclusion, the data discussed support the hypothesis that IL-23 is a central driver of psoriasis. Studies show that blockade of IL-23 with guselkumab is associated with a clinical response, a normalisation of the psoriasis transcriptome, and a reduction in inflammatory cytokines of the IL-23/IL-17 pathway, such as IL-17A, IL-17F, and IL-22.
The Role of IL-23: From Disease Control to Disease Remission
Doctor Lluís Puig
Since the 1980s, it has been recognised that T cells are implicated in psoriatic disease, but the role of IL-23 only began to gain prominence in 2004.15 In the current model of psoriasis pathophysiology, environmental stress causes keratinocytes to produce primary cytokines that activate antigen-presenting cells (usually dendritic cells), which then produce IL-23. In turn, via the IL-23 receptor (IL-23R) expressed on their surfaces, Th17 cells are stimulated to produce IL-17, which leads to the release of various cytokines that promote local keratinocyte activation, epidermal remodelling, and psoriatic plaque formation.15 Therefore, the main rationale for blocking IL-23 in psoriasis treatment is to prevent the IL-23/Th17-mediated ‘feed-forward’ mechanism, which self-amplifies the inflammatory response in keratocytes of psoriatic skin.7 Hence, blockade of the upstream regulator (IL-23) rather than the effector (IL-17) cytokine may be a more effective approach to psoriasis control. This question is currently being addressed in clinical trials involving a range of mAb that block either IL-23 or IL-17, with the latter group requiring a relatively high frequency of dosing in maintenance treatment to be effective.
Another possible advantage of IL-23 blockade is that the effects are not limited to targeting Th17. For example, the effects of IL-23 on regulatory T cells may promote differentiation into Th17 cells,16 as well as affecting cell types known to be present in the skin, such as mast cells, which may be stimulated to promote extracellular trap formation and degranulation, and neutrophils.17 As discussed, a localised disease memory, in the form of epidermal Th22 and Tc17 cells, can form in cases of clinically healed psoriasis. In this setting, epidermal CD8+ T cells are activated and a proportion become enriched in tissue that has healed, including those that express IL-23R as well as cutaneous lymphocyte-associated antigen, CCR6, and CD103.11 These CD8+ T cells respond to ex vivo stimulation by producing IL-17A, while epidermal CD4+ T cells respond by producing IL-22 for as long as 6 years following TNF-α inhibition.11 These pathways have the potential to be modified by agents that target IL-23.
Other clinical advantages of blocking IL-23 include differential impacts on the bowel mucosa important for inflammatory bowel disease (IBD), a reduced risk of candidiasis or other opportunistic infection versus the risk with blockade of IL-17, and potential impacts on neoplasm formation. In the gut, unlike other tissues such as the skin, IL-17 promotes homeostasis and tissue repair rather than driving pathogenic inflammation; nevertheless, it is clear that antibodies targeting IL-23 ameliorate IBD. Data from a mouse model of IL-17A-producing gut cells suggest that the activity of these cells is independent of IL-23, implying that antibodies against IL-23 would not impair IL-17 production by innate lymphocytes. These data help to explain the observation that targeting IL-17 is ineffective in IBD.18 In opportunistic infections of the mucosa caused by Candida albicans, IL-17 signalling is key to immunity and absence of the IL-17 receptor (IL-17R) in mice or humans leads to chronic infection;19 therefore, blockade of IL-23 may represent an alternative therapeutic strategy. More generally, the marked redundancy seen in pathways involved in the IL-effector response to a wide range of pathogens suggests that IL-12/23 blockade should not have a significant impact on signalling, implying a favourable safety profile for IL-23 targeted agents (Figure 1).20 Finally, in immune surveillance, IL-12 acts on lymphoid cells, such as natural killer cells and CD8+ cytotoxic T lymphocytes, which then produce IFN-γ and prevent tumour initiation, growth, and metastasis. In mouse tumour models, there is evidence for various activities of IL-23 in disease: as a tumour suppressor in ultraviolet-induced skin cancer, as an inducer (when overexpressed) of de novo intestinal tumours, and as a target for eliminating residual tumour cells from occult tumours.6 Ultimately, head-to-head clinical trials will determine the extent of the advantages in blocking IL-23 versus IL-17A. Several such clinical trials are currently ongoing in patients with psoriasis.
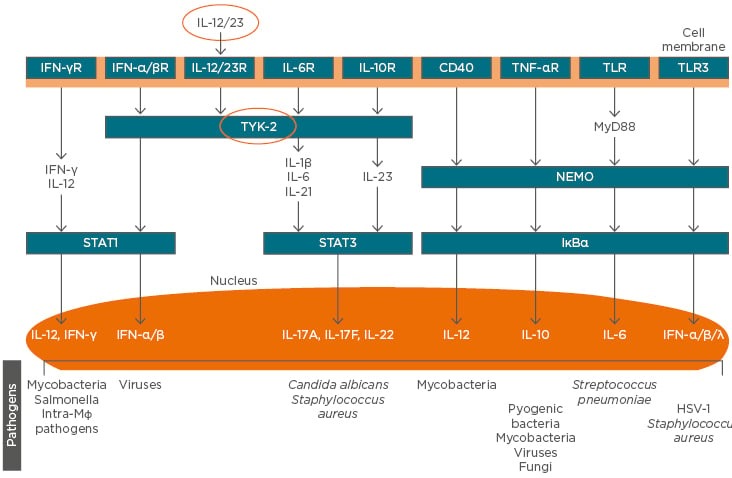
Figure 1: The role of cytokines in the pathogenesis of psoriasis and immune defence against infectious agents, showing redundancy in pathways downstream of IL-12/23 in Th cells that may favour the targeting of regulatory, rather than effector, cytokine blockade in the avoidance of infection.
HSV: herpes simplex virus; NEMO: NFκB essential modulator; R: receptor; TLR: toll-like receptor; TYK: tyrosine kinase.
Adapted from Blauvelt et al.20
Cellular and Molecular Changes in Response to Selective IL-23 Versus Dual IL-12/23 Blockade in Psoriatic Skin
Professor James Krueger
The two founding members of the IL-12 cytokine family, IL-12 and IL-23, share a common p40 subunit but are distinguished by their unique p35 and p19 subunits and their predominant downstream activity of IFN-γ or IL-17 activation, respectively.6 The accepted disease model in psoriasis was, until less than a decade ago, one of inflammatory dendritic cells stimulating keratinocytes to produce a range of multiple cytokines, chemokines, and other inflammatory molecular and cellular effects that resulted in lesion formation, plus feedback and perpetuation of this reaction.21 However, with the more recent availability of specific antibodies to p40 (e.g., ustekinumab) and p19 (e.g., guselkumab), the pathogenic axis was more specifically recognised as IL-23/IL-17, and the respective clinical effects of these differentially targeted mAb have generated much discussion and research interest.
As noted earlier in the symposium, data from head-to-head studies of guselkumab and ustekinumab are lacking. However, biopsy data comparisons have been made using samples from individuals treated in separate clinical trials of the two agents: the Phase III ACCEPT (T12)22 study, combining patients treated with high-dose guselkumab 100 mg and 300 mg, and the Phase I study,23 in which patients were treated with ustekinumab 90 mg. The two patient cohorts shared similar characteristics, with comparable baseline demographics, disease characteristics, and skin histopathology, and all the samples were fed into identical analyses.24 Expression analyses indicated that >2,900 gene transcripts were upregulated in psoriasis lesion tissue, but in ‘recovered’ tissue 12 weeks post-treatment, a higher rate of renormalised (i.e., modulated ≤2-fold) transcripts was seen in those treated with guselkumab (77%) versus ustekinumab (45%) (unpublished data). Also, 75% of transcripts returned to a baseline level ≥75% of normal with guselkumab treatment, versus only 27% with ustekinumab. A ‘molecular scar’ can be identified at Week 12 of treatment versus baseline, in which the transcriptome recovers to 17% of its previous value with guselkumab, versus 58% with ustekinumab. After both 1 and 12 weeks of treatment, the neutralisation of activity of relevant transcriptomic genes following high-dose guselkumab was significantly more extensive than that with ustekinumab (unpublished data). These data were further supported in a real-time PCR analysis of the DEFB4 and LCN2 gene products, showing that these IL-17-responsive antimicrobial proteins recovered to a greater extent with guselkumab versus ustekinumab.24 Histological staining of tissue using markers for keratin 16, T cells, dendritic cells, and other markers also demonstrated 12-week recovery with ustekinumab. These observations prompt the question of the relative potency of guselkumab and ustekinumab, and data show that, across a range of assays, there is a 2–14-fold difference in potency in favour of guselkumab.24
There are several factors that could contribute to the superiority of guselkumab over ustekinumab in neutralising psoriasis-related gene expression. In a mouse model of IL-17-mediated inflammatory activity in skin, knockout of the IL-12 subunit p40 resulted in inflammation, thin skin, and a doubling in transepidermal water loss.5 Therefore, IL-12 may counter-regulate the IL-23/Th17 axis, which is critical for sustaining psoriasis. In addition, there is complexity within the IL-12 family of cytokines, and gene expression data reveal a possible role for other, less well-characterised members. As well as changes in the levels of various members of the IL-12 family, such as K16, IL-17A, p19, and p40, psoriasis is also associated with raised IL-27 (unpublished data). IL-27 is composed of the subunits p28 and Ebi3 (named for homology to an Epstein–Barr virus gene),25 neither of which are targeted by guselkumab or ustekinumab. As the IL-12 cytokine family is promiscuous and protein subunits of the family can combine with different partners to activate other pathways, Ebi3 could pair with p19 to form IL-39.25 In a mouse model of lupus, IL-39 drives inflammation, including neutrophil activation,26 and although a native human IL-39 has not been identified, the subunits are both elevated in psoriasis cells (unpublished data). Moreover, p40 can pair with p28 to form IL-Y,7 which has anti-inflammatory activities; therefore, it is possible that some of the benefits of blocking IL-12 and IL-23 activity could be reduced by downregulating beneficial IL-Y activity. Furthermore, there may be functional plasticity in the Th17 lineage, such that removal of IL-23 from pathogenic T cells can convert them to non-pathogenic, regulatory T cells.27 Any or all of these effects may play a role and require further investigation. In summary, although molecular data have shown very clear differential effects of guselkumab and ustekinumab on the transcriptome of psoriasis-associated cells, other potential cytokine activities in psoriasis still require full characterisation.
Disease Modification in Psoriasis: Fantasy or Reality?
Doctor Curdin Conrad
In patients with psoriasis receiving anti-IL-23 treatment, a positive response to continuous treatment can be very long-lasting. A high rate of freedom from disease has been seen with continuous guselkumab treatment in the Phase III VOYAGE studies8,12 and with risankizumab in a Phase II study.28 This clinical benefit is beyond that anticipated based on the half-life of the drugs and raises the possibility that, by some mechanism, a form of disease modification has resulted from treatment. Such a mechanism may involve activated T cell migration to the lymph nodes, where they perform a central memory function and/or reside in the skin for a long period. As noted, evidence for the latter originates from disease memory in clinically healed skin, which shows relatively high levels of IL-17-producing T cells.29 It has been proposed that, following successful treatment, the in situ activation of epidermal T cells resident in psoriatic skin can lead to IL-17A production, resulting in recruitment of further inflammatory T cells from the blood and subsequent clinical relapse.12 This suggests that, to have any long-term disease-modifying effect, skin-resident memory T cells should be targeted.
In psoriasis, Th17 and Tc17 cells coproduce IL-17A and other cytokines, with their expansion dependent on IL-23.30,31 The physiological function of these cells is thought to be protection from extracellular pathogen attack (Figure 1); however, overexpression in autoimmune disease is also common.30 It is clear that to achieve a response to psoriasis treatment, a reduction of IL-17 levels is necessary,32 and with the array of targeted agents available (e.g., TNF inhibitors, IL-12/23 or IL-23 inhibitors, IL-17A or IL-17R inhibitors) there are many methods to achieve this. Relapse following discontinuation of IL-17R blockade generally occurs within a few weeks,33 suggesting that blockade of IL-17 rather than its receptor may be a more efficacious long-term approach. In Crohn’s disease, in which IL-17 is highly expressed, expectations for anti-IL-17 treatment were not fulfilled; indeed, cases of aggravated IBD following anti-IL-17 treatment were observed.30,34 One explanation for this is the existence of two types of IL-17-producing cells: pathogenic Th17 cells and non-pathogenic Th17 cells that also produce IL-10 (which also provide a beneficial barrier and pathogen defence function) independent of IL-23 signalling.35 Only the former are blocked by IL-23 targeting.
How can we effectively assess the effects on IL-17 and IL-22-producing skin-resident memory T cells present in non-lesional tissue? Following treatment discontinuation, psoriasis tends to revert to its baseline severity.11 In a recent study of secukinumab treatment discontinuation, gene expression analysis of non-lesional skin in patients who did not relapse showed a robust, durable effect 1 year after stopping therapy.36 This may be due to the removal of memory T cells from non-lesional skin. From hypotheses about any long-term effects on these cells, it has been suggested that targeting IL-23 may be beneficial in preventing Th17/Tc17 cells from becoming pathogenic. In addition, single nucleotide polymorphisms in IL-23R are associated with autoimmune disease, including psoriasis, and IL-23R is preferentially expressed in these skin cells in psoriasis patients. Possibilities for purging memory T cells include antibody-dependent cell-mediated cytotoxicity or lack of stimulus through IL-23R, though evidence for any of the available drugs exerting either mechanism in non-lesional skin is lacking. In conclusion, multiple observations suggest disease modification in patients with psoriasis receiving anti-IL-23 treatment: a clinical effect beyond drug half-life and biological half-life of the treatment, possible effects on skin-resident memory T cells that mediate disease memory, and the effect of blocking only pathogenic (not non-pathogenic) Th17 cells.
Click here to view the full symposium.




