
Abstract
Stevens-Johnson syndrome and toxic epidermal necrolysis are among the most concerning drug reactions affecting adults and children. Although the overall mortality has reduced substantially after the introduction of several strategies, such as prompt withdrawal of the causal drug and management of the patients in an intensive care or burn unit, these conditions continue to be associated with severe complications and a mortality rate of 1–4%. Currently, several treatment options including systemic corticosteroids, intravenous immunoglobulins, cyclosporine, tumour necrosis factor-α inhibitors, and plasmapheresis among others, have shown inconclusive benefits regarding their efficacy and safety in patients with these conditions. This review analyses the most recent literature regarding treatment options for paediatric patients with Stevens-Johnson syndrome and toxic epidermal necrolysis.
INTRODUCTION
Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN) are two uncommon but widely concerning conditions that affect both children and adults worldwide. Their overall annual incidence has been reported in 1–10 cases/1 million, of which 20% are paediatric cases.1 Mortality rates in SJS range between 1% and 4%, whereas in TEN it increases to between 25% and 35%, being somewhat lower in children.2 SJS/TEN represent entities in the same disease spectrum with different degrees of severity.3,4 Recently Roujea5 suggested denominating these conditions as epidermal necrolysis because both are characterised by skin and mucosal detachment due to keratinocyte apoptosis. SJS/TEN are mostly triggered by drugs such as anticonvulsants, allopurinol, sulfas, and antibiotics, and less frequently by infections such as mycoplasma pneumonia. However, other conditions have also been associated.6 Although the precise pathogenesis of SJS/TEN remains uncertain, it is considered that specific agents (e.g. drugs, infection) elicit an immune-mediated cytotoxic reaction against keratinocytes, generating extensive apoptosis. The main cytotoxic molecules involved in its mechanism are granulysin, perforin/granzyme B, and Fas ligand.7 More recently, the role of T helper 17 cells in the pathogenesis of SJS/TEN as enhancers of the immune response in affected patients has been proposed.8 Additionally, several reports have shown a strong association among some HLA genes, with SJS/TEN incidence, as a result of specific drugs such as carbamazepine (HLA-B*58:02, HLA-A*31:01) and allopurinol (HLA-B*58:01).5,9
Clinically, the SJS/TEN spectrum is divided into three groups based on the body surface area (BSA) involved. SJS affects <10% of the BSA, while TEN affects >30%. SJS/TEN overlap refers to a skin and mucosae involvement between 10% and 30% of BSA.8 Patients may present a prodromal stage (fever, cough, sore throat, and general malaise) followed by skin and mucosal lesions. The skin lesions begin as erythematous macules with a possibility of developing rapidly into papules, vesicles, bullae, urticarial plaques, or confluent erythema and when the bullous lesions break, they leave large areas of denuded skin. Other lesions may have a peculiar appearance of ‘flat atypical targets’ differing from the typical target lesions of erythema multiforme (Table 1). At least two mucosae are always affected to a lesser or greater degree, and may present with erythema, oedema, sloughing, blistering, ulceration, and/or necrosis. Extensive loss of the epidermis layer leads to infections, electrolyte imbalance, and in some cases organ failure that could result in death.10
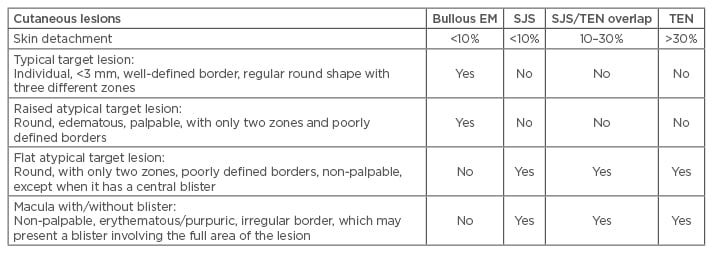
Table 1: Clinical features of erythema multiforme, Stevens-Johnson syndrome, and toxic epidermal necrolysis.
EM: erythema multiforme; SJS: Stevens-Johnson syndrome; TEN: toxic epidermal necrolysis.
Modified from Bastuji-Garin et al.3
Although laboratory findings are not specific, several haematological or biochemical parameters can show abnormalities, including liver function and renal function tests.1 Skin biopsies are very useful to rule out other entities (e.g. fixed drug eruption, Staphylococcal scalded skin syndrome, and other bullous diseases). They usually show varying degrees of vacuolisation of the basal membrane, including sub-epidermal blisters and keratinocyte apoptosis, possibly progressing to full-thickness necrosis. Sparse perivascular lymphoid infiltrate is also characteristically present.10,11
The severity of illness score for TEN patients (SCORTEN) was described by Bastuji-Garin et al.3 The SCORTEN predicts the risk of death and evaluates different parameters at the time of hospital admission including: age >40 years, malignancy, heart rate >120/min, initial percentage of epidermal detachment >10%, blood urea >28 mg/dL, serum glucose >252 mg/dL, and serum bicarbonate <20 meq/L. The score ranges from 0 to 9; a score ≥5 is associated with a risk of death of 90%.12 Long-term sequelae and recurrence may present in ≤47% of patients. Sequelae include post-inflammatory hypo/hyperpigmentation, scarring, uveitis, keratitis, corneal defects, blindness, sclerosing cholangitis, bronchiolitis obliterans, stridor, and venous thrombosis, among others. The prognosis depends on age, the percentage of BSA affected, and the associated comorbidities.13 The treatment options more frequently used worldwide include intravenous immunoglobulin (IVIg) and systemic corticosteroids. Recently, cyclosporine (CsA) and tumour necrosis factor-α (TNF-α) inhibitors have also been explored in these conditions.14-19 This manuscript reviews the recent literature regarding treatment options for the paediatric population.
TREATMENT UPDATE
Supportive Therapy
The initial management of SJS/TEN consists of the early withdrawal of the responsible drug and supportive therapy (ST) in an intensive care or burn unit, including fluid resuscitation, nutritional support, and the prevention of infections and sequelae.4 Even though the overall mortality in patients with SJS/TEN has been reduced substantially thanks to advances in ST, a recent systematic review showed that patients receiving only ST took longer to achieve remission, had longer hospital stays, and presented more complications and deaths compared with patients treated with systemic therapy, such as systemic steroids or IVIg.14
Systemic Corticosteroids
Corticosteroids decrease the synthesis of pro-inflammatory molecules and inhibit prostaglandin and leukotriene production. These drugs also have anti-proliferative effects and impair monocyte and lymphocyte function. In theory, their mechanism of action could modify the uncontrolled immune response observed in patients with SJS/TEN though their role in the management of SJS/TEN patients remains questionable. For some authors, the use of systemic corticosteroids increased the risk of complications, infections, and hospital stays, and should thus be avoided.20 On the other hand, others proclaim the benefits of steroid use in these conditions, such as a reduction in the duration of fever and skin eruption.21 The multinational study EuroSCAR evaluated the role of corticosteroids in adults with SJS/TEN. The study found that prior use of corticosteroids in patients with SJS/TEN prolonged the period of disease progression but did not influence the disease severity or mortality.22 While similar studies in paediatric patients have not yet been performed, we have recently published a systematic review evaluating the outcome of several treatment modalities for drug induced SJS/TEN in children, showing that patients with SJS receiving systemic corticosteroids (prednisone, prednisolone, or methylprednisolone) had a better outcome than patients receiving ST alone. Although in both groups the percentage of complications were similar (25%), the severity was higher in the ST group (e.g. sepsis, death) compared with the corticosteroid group (e.g. scarring, hyperpigmentation, mild infections, bronchiolitis obliterans).14
More recently Finkelstein et al.13 reported the outcome of 55 retrospective paediatric cases of SJS/TEN. They found that patients with no exposure to corticosteroids had a higher association with ocular sequelae than patients treated with these medications. Additionally, a retrospective study performed in Thailand reviewed 189 paediatric cases of SJS/TEN treated between 1979 and 2007, in which patients were divided into three groups depending on the period in which they were admitted to the hospital. Overall, 58% of the patients received treatment with systemic corticosteroids. The authors reported that the use of corticosteroids in their institution increased progressively from 18% to 64% to 87% (first, second, and third periods, respectively) due to the positive outcomes of these patients. Complications in the three groups reached 20% and included infections, eye sequelae, and hepatitis among others. Interestingly, and contrary to previous reports in the literature, the mortality decreased from 9% to 1.5%, this being lower during the last period precisely when more patients received steroids, although this could also have been associated with advances in supportive care in more recent years.23 Ferrándiz-Pulido et al.24 similarly reported a review of 14 paediatric cases of SJS/TEN of which 86% received treatment with systemic corticosteroids. The overall mortality rate in this study was 7%, but no deaths were reported in the corticosteroid group.24 Despite the above data potentially suggesting the potential benefits of corticosteroids in paediatric patients with SJS/TEN, it is important to mention that there is no consensus regarding dose, type of corticosteroid given, and length of treatment for these patients, and in most of the studies these vary greatly.
Intravenous Immunoglobulin
IVIg consists of exogenous pooled human immunoglobulins, mainly IgG (IgG 1–4) and a minimal supply of IgA, IgE, and IgM antibodies, including autoantibodies against ubiquitous proteins such as Fas. The mechanism of action of IVIg is complex and still not completely understood, however its use in SJS/TEN has become popular following the in vitro demonstration by Viard et al.,25 showing that IVIg can block the binding between apoptotic Fas ligand (CD95L) and its respective apoptotic receptor, located on the keratinocyte cell surface, responsible for the programmed cell death observed in patients with these conditions.25 Initial data indicated that IVIg was superior to systemic corticosteroids in the management of severe drug reactions,26 however further studies have shown contradictory results.22,27 In 2008, the EuroSCAR study reported that adults with SJS/TEN treated with IVIg had no better outcomes compared with other treatment modalities (e.g. systemic corticosteroids, ST); moreover, the mortality was higher in this group.28 However, some authors note that the IVIg group (EuroSCAR study) had a higher proportion of patients with TEN versus SJS, than other groups. They also suggest that many of these patients did not receive IVIg before Day 4 as recommended, that they were not admitted to a burn unit, and that they may have received sucrose-containing IVIg, which has been associated with renal toxicity.29 Thus, several authors still consider that IVIg is the best treatment option for patients with SJS/TEN.
A recent survey performed among North American physicians with a special interest in patients with SJS/TEN showed that >70% of them preferred to use IVIg in patients with diagnosed TEN and >50% use IVIg in at least one form of SJS (SJS or SJS/TEN overlap).19 On the other hand, our systematic review did not show a statistically significant difference in the outcome among patients who received systemic steroids versus IVIg.14 More recently, Barron et al.6 performed a meta-analysis with meta-regression of observational studies regarding IVIg in the treatment of SJS/TEN (both adults and children). They concluded that high doses (>2 g/kg) seemed to significantly decrease mortality in these patients, however other outcomes such as time of stay, sequelae, and other complications were not evaluated.6 Finkelstein et al.13 reported a retrospective study of 55 paediatric cases of SJS/TEN, in which 38% of the patients received IVIg as treatment (21% of them also received concomitant treatment with systemic corticosteroids). Interestingly, the group of patients treated with IVIg had a higher incidence of ocular complications.13
Despite literature showing that IVIg is relatively safe, it has an overall risk of adverse reactions of 10%, and although most of them are minor (mainly infusion reactions), serious adverse reactions like renal failure, aseptic meningitis, stroke, infection, haemolysis, deep venous thrombosis, and anaphylaxis have been reported.30 Infusion reaction symptoms include headache, nausea, fever, vomiting, cough, malaise, myalgia, arthralgia, abdominal pain, flushing, urticarial lesions, and variations in heart rate/blood pressure.31 Similarly to corticosteroids, guidelines regarding dosage and length of treatment vary widely among authors.2 Finally, some authors consider that a combination of corticosteroids plus IVIg has a superior therapeutic effect and a reduced mortality associated in patients with SJS/TEN compared with any of these two medications alone, however evidence needs to be reviewed in more detail.12,32,33
Cyclosporine
CsA is a powerful immunosuppressive and immunomodulatory drug, used traditionally to prevent rejection of transplanted organs and other inflammatory or autoimmune conditions such as pemphigus, atopic dermatitis, and psoriasis, among others.34 CsA targets mainly T cell-dependent immune mechanisms, which are implicated in transplant rejection and some forms of autoimmunity, through the inhibition of T helper cells and cytotoxic T cells. It also selectively blocks many immunoregulatory functions of activated T cells, thereby inhibiting the release of interleukin (IL)-3, IL-4, IL-5, interferon-g, granulocyte monocyte colony stimulating factor, and TNF-α.35
The use of CsA in the treatment of SJS/TEN was first reported almost two decades ago. In 2000, Arévalo et al.36 reported 11 adult patients treated with oral CsA twice daily (3 mg/kg/day) and then compared them with a historical series of patients (same institution) treated with either cyclophosphamide or different doses of corticosteroids. The authors found that the patients treated with CsA had an early time of arrest of disease progression and a shorter time of re-epithelialisation in comparison with the other patients.36 Following this, other case series and case reports have also shown the potential benefit of CsA in the management of patients with SJS/TEN, although unfortunately, studies including children are scarce.16,37,38 Aihara et al.39 reported a case of a child with TEN treated successfully with IV CsA (1 mg/kg/day) and methylprednisolone (30 mg/kg/day), showing clinical and laboratory improvement in the first 24 hours after starting treatment. Valeyrie-Allanore et al.40 performed an open trial of CsA in 29 patients with SJS/TEN, of which only 3 were <19 years old. All the patients were treated with oral CsA solution (through nasogastric tube) with an initial dose of 1.5 mg/kg twice daily for 10 days, with subsequent tapering until completing 1 month of treatment. The authors found that both mortality and the progression of detachment were lower than expected, and only a few patients presented complications (n=3) (leukoencephalopathy, neutropenia, and nosocomial pneumopathy). In two other patients, the dose was reduced early due to renal impairment.40
More recently Singh et al.18 performed a retrospective comparison among 11 patients with SJS/TEN, treated with CsA, and patients treated with systemic corticosteroids for the same conditions. In this study, only two children were included (a 14-year-old and a 7-year-old). Overall the mean duration of re-epithelialisation was 14.5 days in the CsA group and 23.0 days for the corticosteroid group. The mean hospital stay was also lower in the CsA group (18 days) compared with the other (26 days). There was no mortality in the CsA groups and there were two mortalities in the group treated with corticosteroids.18 Although the evidence suggesting that CsA could have a potential role in the management of paediatric patients with SJS/TEN is limited, the experience in other conditions such as atopic dermatitis and psoriasis may encourage other health professionals to develop better quality studies in the future and thus properly establish the use of CsA in the treatment of children with SJS/TEN.
Plasmapheresis and Haemoperfusion
Plasmapheresis and haemoperfusion are well-recognised procedures, characterised by the removal of toxic or pathological molecules from the blood potentially contributing to disease progression. Both procedures are currently used in children in the treatment of several inflammatory and/or immunological conditions, such as sepsis, Guillan-Barré, Henoch-Schönlein purpura, recalcitrant atopic dermatitis, and pemphigus vulgaris, among others.41,42 Although the role of plasmapheresis/haemoperfusion in the treatment of paediatric SJS/TEN has not been well established, it can offer a potential benefit principally for patients with severe disease (mainly TEN) or those who have not responded to other treatments (corticosteroids and/or IVIg),43 and is currently a Class III indication of the American Society for Apheresis (ASFA).42 The mechanism by which plasmapheresis/haemoperfusion appear effective is by removing drugs and their metabolites, or immune complexes and inflammatory mediators that have been released and promote keratinocyte apoptosis, from the blood of the patient.44
The use of plasmapheresis in patients with TEN was initially reported in the 1980s and later, several authors also reported favourable outcomes, although these studies mainly included adults.45 Subsequently, Chaidemenos et al.,46 Egan et al.,47 and Koštál et al.48 published three independent case series including patients with TEN, which were successfully treated with plasmapheresis. However, only six paediatric patients were reported in these studies (two in each study).14,48 More recently, Hinc-Kasprzyk et al.43 reported a 4-year-old boy with late-stage TEN, who had previously failed systemic therapy with corticosteroids and IVIg, that was treated with plasmapheresis. The patient showed a remarkable improvement of his general condition only 2 days after the second plasmapheresis session.43 Some of the disadvantages of plasmapheresis include its high cost and its risk of blood transfusion-related side effects, such as citrate effect, decrease in blood pressure, and allergic reactions.44,48 Furthermore it is not widely available, particularly in resource-constrained environments.49
Haemoperfusion, on the other hand, does not seem to have the same limitations, as this requires less costly equipment and is easier to perform compared to plasmapheresis.44 Its success in the management of patients with sepsis has raised the interest of some authors to adapt this procedure as a new treatment option for patients with severe SJS/TEN. Wang et al.44 reported in 2014 a series of seven paediatric patients with SJS/TEN treated satisfactorily with this procedure. All patients received a number of sessions, ranging from 3–5, and showed a rapid improvement of symptoms (fever or progression of the skin rash) after the first session. Patients’ skin lesions healed after 7.0 days and the average length of hospital stay was 14.4 days. Some adverse reactions observed in these patients included hypotension and palpitations, and three patients developed femoral vein thrombosis which resolved with anticoagulant treatment.
Tumour Necrosis Factor-α Inhibitors
Biological agents specifically target mediators of inflammation by stimulating or suppressing particular components of the immune response. In children, TNF-α etanercept, infliximab, and rituximab are the most prevalent agents currently used to treat a wide variety of autoimmune and inflammatory conditions (psoriasis, atopic dermatitis, inflammatory bowel disease, juvenile arthritis).50 Recent literature has showed high levels of TNF-α in skin biopsies of patients with TEN prompting several authors to use some of these agents in the management of patients with SJS/TEN.51 In children, few reports have been published (three patients with infliximab and one with etanercept), and although the results seem encouraging, their use needs to be cautiously considered due to the potential side effects (increased risk of infections, haematological abnormalities, lymphoma, hepatotoxicity) and elevated costs compared with other treatment modalities (CsA or methylprednisolone) (Table 2).50 Other drugs with similar anti-TNF-α properties, but with relatively low costs, include N-acetylcysteine and pentoxifylline. Few cases of paediatric patients with SJS/TEN being successfully treated with these agents have been reported thus far and unfortunately the literature regarding this topic remains limited.10,14,52
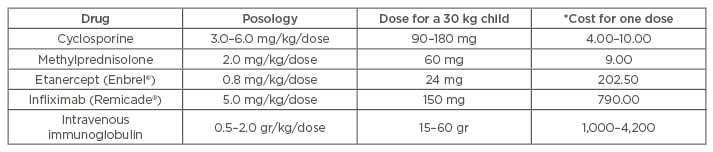
Table 2: Estimated cost of different treatment modalities for Stevens-Johnson syndrome/toxic epidermal necrolysis in the province of Ontario, Canada.
*Costs are current as of 2015 and expressed in Canadian dollars. They represent exclusively the cost of the drug/agent and do not include administration costs such as nursing. Costs may vary depending on the country and healthcare system.
Costs obtained from www.transfusionontario.org and www.health.gov.on.ca.
CONCLUSIONS
In spite of the current variety of treatment options reported in the literature regarding the management of SJS/TEN in children, there is not enough reliable evidence to establish their safety and efficacy, as randomised controlled trials are difficult to perform, mainly due to the rarity of this disease. However, better quality studies could be conducted in the future through international collaborative programmes or registries that establish standardisation of inclusion criteria, clinical and pathological information, outcome definitions, and treatment protocols based on expert consensus.





