Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant hereditary disease with hamartomatous growths in multiple organs due to loss-of-function variants in TSC1 or TSC2. In approximately 15% of patients with clinical TSC, no pathogenic variant can be identified, and low-level mosaicism is suggested to be one of the reasons. Mosaicism is well-known in TSC and challenges the molecular genetic diagnosis. The advent of next-generation sequencing has improved the diagnostics in TSC including in patients with mosaicism. The TSC phenotype varies widely, and mosaic patients with TSC are often considered to have a milder phenotype. Here, the authors describe a patient with mosaic TSC with a 10% variant allele fraction and manifestations in three organ systems (skin, eyes, and kidneys). Furthermore, the authors studied existing literature about phenotypic organ manifestations in patients with mosaic TSC. No clear definition of the phenotype of patients with mosaic TSC could be established, but unilateral angiofibromas and the absence of tubers and a subependymal nodule could indicate mosaicism. The case shows that patients with low-level mosaic TSC can have multiple affected organ systems though still a mild clinical picture.
INTRODUCTION
Tuberous sclerosis complex (TSC) is an autosomal dominant hereditary disease characterised by a variable phenotype and growth of hamartomas in multiple organs.1-6 Affected organs include skin, brain, eyes, heart, kidneys, and lungs, and the diagnosis is often established in childhood, where most patients develop seizures.7 The disease is caused by the pathogenic loss-of-function variants in the tumour suppressor genes TSC1 and TSC2.5,6,8,9 In patients with clinical TSC, pathogenic variants of TSC1 and TSC2 can be identified in a blood sample in 75–90% of cases.3 One-third of patients have an inherited form, while the other two-thirds result from a de novo variant.1,2,10
Mosaicism in TSC arises from post-zygotic mutations in TSC1 and TSC2, whereby the cells in an individual harbour different genotypes (Figure 1).2,4,5,11 It can be generalised mosaicism manifesting in several cell types and organs or mosaicism limited to a single cell line.11,12 Mosaicism challenges the molecular genetic diagnosis of TSC.2 In approximately 15% of patients with clinical TSC, no pathogenic variant can be identified in the blood (no mutation identified [NMI]).1-3,13 Mosaicism with a low variant allele fraction (VAF; mosaic variant reads/total reads) is one of the reasons and is suggested to constitute approximately 50% of NMI cases.1,3,12-14
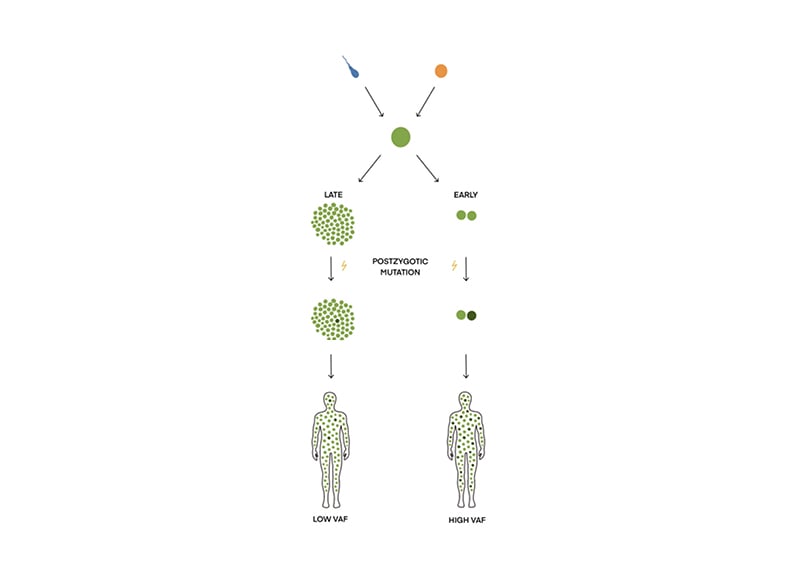
Figure 1: How mosaicism arises during embryogenesis.
Mosaicism is the presence of two or more cell populations with different genotypes. It arises from a single zygote. If the post-zygotic mutation happens early during embryogenesis, the VAF (mosaic variant reads/total reads) will be high; if the mutation occurs late, VAF will be low.
VAF: variant allele fraction.
Here, the authors present their most recent case of mosaic TSC in a 32-year-old male, who was diagnosed with low-level mosaicism and several affected organs. They provide a review of published literature regarding mosaicism in TSC to elucidate the phenotype in patients with mosaic TSC. Furthermore, original data from Danish hospitals are presented.
CASE REPORT
A 32-year-old male was referred to the Department of Clinical Genetics to confirm a clinical diagnosis of TSC. At the age of 11 years, he had an angiofibroma removed from his chin. Since then, several skin lesions had emerged around his nose and chin. He was the only child of seemingly healthy parents and there were no other cases of TSC known in the family. He had no history of seizures. A diagnostic work-up with an MRI scan of the cerebrum was unremarkable. An MRI scan of the abdomen showed multiple small cysts in both kidneys as well as two larger angiomyolipomas, which were confirmed by a CT scan and an ultrasound-guided biopsy. A dermatological evaluation revealed numerous angiofibromas in the central part of the face and three hypomelanotic macules on the left forearm, left lower part of the back, and on his left leg (Figure 2). An ophthalmologist could not rule out or confirm TSC when examining the eyes. A molecular genetic analysis using standardised next-generation sequencing (NGS) with the HaloPlex Target Enrichment System (Agilent, California, USA) and data filtering, in which all variants observed with an allele frequency of 10% or less were removed, showed no pathogenic variant in the TSC1 or TSC2 gene. At this point, a mosaic form of TSC was suspected. A revised, more sensitive version of the molecular genetic analysis of TSC1 and TSC2 (HaloPlex™ HS Target Enrichment System; Agilent) and data filtering, in which only variants observed with an allele frequency of 1% or less were removed, showed a pathogenic splice variant in the TSC2 gene (c.4663-1G>A) in a mosaic form (10%). A new evaluation by the Department of Ophthalmology showed a suspected astrocytoma in the right eye.
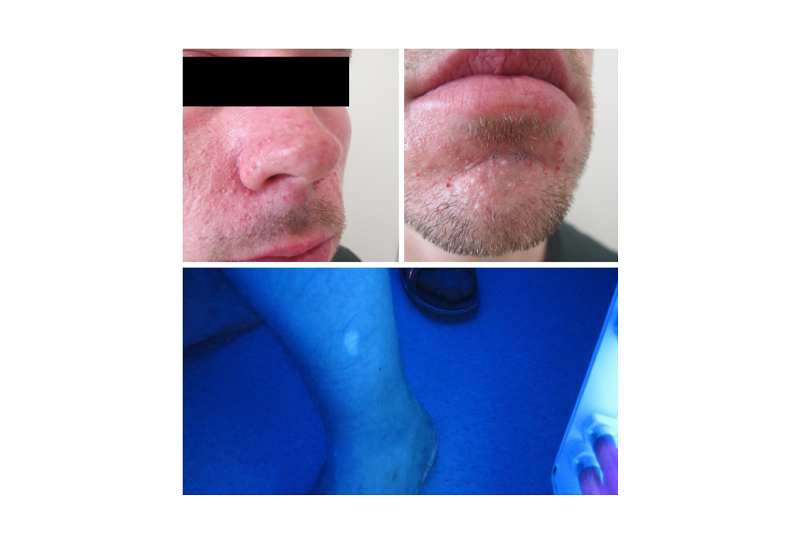
Figure 2: Patient with mosaic tuberous sclerosis complex, symmetric facial angiofibromas, and hypomelanotic macula illustrated in Woods light.
METHODS
At the Department of Clinical Genetics, Copenhagen University Hospital, Rigshospitalet, Denmark, the coding regions of TSC1 and TSC2 were sequenced by NGS using a MiSeq benchtop sequencer (Illumina, San Diego, California, USA) after HaloPlex™ Custom Target Enrichment System (Agilent). The obtained data were subsequently analysed, and the BAM and VCF files were obtained using SureCall NGS Data Analysis software (Agilent), with the help of the Burrows-Wheeler Alignment maximal exact matches tool and SNPPET single nucleotide polymorphism caller. At least 99% of the target regions (exons including 20 base pair flanking regions) were covered by at least 20 reads.
HaloPlex HS was used, and all variants with an allele frequency of >1 % were analysed. Using this very sensitive method, a splice variant, c.4663-1G>A, was identified in mosaic form (10%). The presence of the variant was confirmed by allele-specific PCR using four different allele-specific primer pairs:
1. Tsc24663-1GWT/mutF: ggtgtgctttgagtgtggag/Tsc24663–1GWtR: tggcgagctcgctgttgctc
2. Tsc24663-1GWtF: gcctctcccctctccccacag/Tsc24663-1GWt/mutR: TGCCAGCAGTAGGTGAACTG
3. Tsc24663-1GWT/MutF: ggtgtgctttgagtgtggag/Tsc24663-1GMutR: tggcgagctcgctgttgctg
4. Tsc24663-1GMutF: gcctctcccctctccccacac/Tsc24663-1GWt/MutR: TGCCAGCAGTAGGTGAACTG
Primer pairs 1 and 2 amplify the normal (wt) allele (G) in a forward and reverse direction, whereas primer pairs 3 and 4 amplify the variant allele (C) in a forward and reverse direction. A normal DNA sample was included as a control in addition to DNA from the authors’ case.
The above is a TSC2 GenBank reference sequence (accession number: NG_005895.1). Numbering is according to the TSC2 GenBank reference sequence (accession number: NM_000548.3), in which nucleotide 1 is A in ATG-translation initiation codon.
DISCUSSION
The case describes a patient with mosaic TSC, with a low VAF variant (10%) but still with three affected organ systems (skin, eyes, and kidneys). This shows that, even though a patient with TSC has a mosaic form of the disease and a mild clinical phenotype, several organ systems can be affected. Regarding the diagnostic criteria of TSC, the patient had three major features (three hypomelanotic macules, facial angiofibromas, and angiomyolipomas) and one minor feature (multiple renal cysts), and thereby fulfilled a clinical diagnosis of TSC (Table 1).15
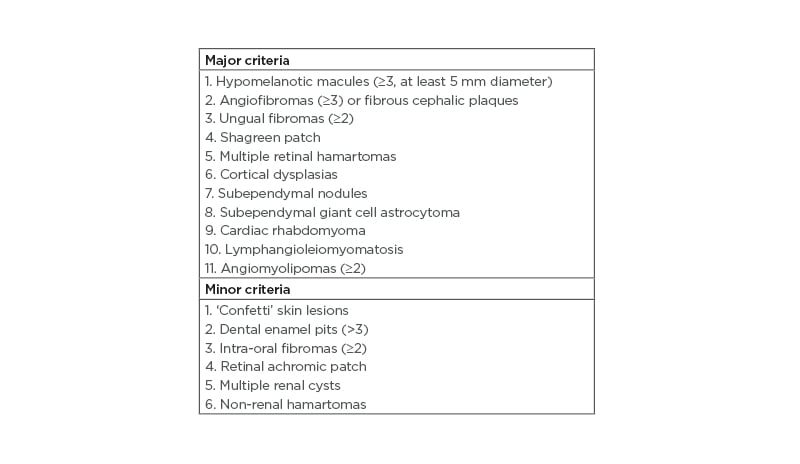
Table 1: Major and minor diagnostic criteria in tuberous sclerosis complex.
Definite diagnosis: two major features or one major feature with ≥2 minor features. Possible diagnosis: either one major feature or ≥2 minor features.15,17
After the introduction of NGS, more pathogenic variants in TSC1 and TSC2 are now detected including pathogenic variants in mosaic state.3,5 NGS, with sufficient depth, can detect pathogenic variants with a very low VAF (>1 %).4,16
Patients with mosaic TSC tend to have fewer major symptoms (Table 1) and fewer organ systems involved compared to patients with germline TSC.2,3 Overall, patients with mosaic TSC have a milder phenotype compared to patients with germline TSC, but the disease spectrum for patients with mosaic TSC can resemble and overlap with patients with germline TSC.1,2,12 Treichel et al.13 have proposed a diagnostic algorithm to detect possible mosaic TSC in patients. It was suggested that low-level mosaicism includes <3 mucocutaneous findings, the absence of tubers and subependymal nodules, and asymmetrical distributed angiofibromas with <100 lesions;13 however, exceptions from this suggestion exist.
Angiofibromas are a common skin manifestation and have been reported in as many as 92% of a cohort of 24 patients with mosaicism.12 Most frequently, angiofibromas are distributed bilaterally over the central part of the face, as with the authors’ patient; however, unilateral involvement has been found in some patients with mosaicism.1,18-20 In the authors’ patient, the hypomelanotic macules were unilateral. Many mucocutaneous findings are less frequent among patients with mosaic TSC, and the time of onset for ungual fibromas and angiofibromas is, in general, delayed.1,12 Overall, there is no significant difference in the prevalence of skin manifestations between patients with germline variants in TSC1 or TSC2 and patients with NMI.4
The neurological phenotype in patients with mosaic TSC can be characteristic and thereby an indication of mosaicism in TSC.1 The most common neurological phenotype in patients with mosaic TSC is the presence of cortical tubers and absence of subependymal nodules. However, the presence as well as the absence of both cortical tubers and subependymal nodules has been observed in patients with mosaic TSC.1,13,21,22 Cortical tubers, subependymal nodules, and subependymal giant cell astrocytomas occur more often in patients with germline TSC compared with patients with mosaic TSC.12 Subependymal nodules and cortical tubers are both relatively frequent manifestations, and subependymal giant cell astrocytomas are more rare manifestations.4,23-26 The authors’ patient had no abnormal findings on his brain scan.
Seizures are a frequent manifestation in patients with germline TSC as two studies have shown 91% and 88% are affected, respectively, while it is less frequently seen in patients with mosaicism (57.0% and 32.3%).12,27
Prevalence of intellectual disability varies greatly in patients with NMI.4,23-26,28-31 The median prevalence value is 39%.4 Both intellectual disability and attention deficit hyperactivity disorder occur more frequently in patients with NMI than patients with a pathogenic variant in TSC1, but less frequently compared to patients with TSC2 variants.4,32 Autism is seen in patients with TSC with a higher prevalence among patients with a TSC2 pathogenic variant, and less frequent among patients with a TSC1 variant and in patients with mosaicism.4,12
Ocular manifestations consist of hamartomas limited to the retina.4,33 Retinal hamartomas have been found in 14.8% of patients with mosaic TSC compared with 21% of patients with TSC, but without statistical significance.12
Renal lesions, including angiomyolipomas and renal cysts, have been described in 39–67% and 18–31% of patients with NMI, respectively.4,23-26,28,34 One study found angiomyolipomas to be more common in patients with mosaicism compared to patients with TSC, although there was no statistically significant difference.12 The TSC2 gene is adjacent to the PKD1 gene, which causes polycystic kidney disease, and a gene alteration involving both genes results in a very serious renal phenotype.4,33,35 Among mosaic patients, deletions involving TSC2 and PKD1 are frequent, but the renal function is better preserved.36
Cardiac rhabdomyomas can be seen in patients with TSC and, less frequently, in patients with mosaicism.12 They have been reported in 33–56% of patients with NMI.4,24-26,28,37
The frequency of pulmonary lymphangio-leiomyomatosis (LAM) in patients with mosaicism has been reported with contradictory results. One study suggests that the prevalence of LAM is the same for patients with mosaicism and patients with germline TSC,1 while another concludes LAM to be less frequent among patients with mosaic TSC.12 Additionally, LAM has been described to occur more often in patients with NMI compared with patients with TSC2 and TSC1 pathogenic variants.4,23,28 The high rate in patients with NMI might be influenced by the small samples that have been analysed.4
When TSC is diagnosed in a family, genetic counselling is necessary. Usually, genetic tests and thorough clinical evaluations of the parents are needed to determine whether the variant is inherited or de novo.4,5 In patients with mosaic TSC, the pathogenic variant has occurred postzygotic and, therefore, is not inherited. The descendants of a patient with mosaicism should always be offered a thorough clinical as well as a genetic examination, since patients with mosaic TSC are at risk of having offspring with non-mosaic TSC.18,38 The risk of inheriting the pathogenic variant is up to 50% in the descendants of patients with mosaic TSC and depends on the size of the VAF.16
If the parents of a child with TSC are unaffected, without a pathogenic variant, and mosaicism has not been confirmed in the index patient, gonadal mosaicism cannot be excluded and the sibling(s) of the patient with TSC should be thoroughly examined.18,38 The incidence of gonadal mosaicism in TSC is approximately 2%.16,18,38
Mosaicism can be defined as a VAF <40%.1,39 The size of VAF depends on the timing of the mutation during embryogenesis (Figure 1).40 VAF varies in different types of tissue within the same patient. Affected skin biopsies have a median VAF that is twice as high as in blood, saliva, and normal skin.3,9,12 The size of VAF and the number of major features correlate, i.e., clinical manifestations can vary depending on the timing of the mutation during embryogenesis. If it occurs early, a large fraction of the cells will contain the pathogenic variant, resulting in a phenotype with more TSC manifestations. If the mutation occurs later, the pathogenic variant will only be present in certain tissues and patients will have a milder phenotype.1,5,18
There is a considerable inter- and intrafamilial variation among organ manifestations.4,5 These make it difficult to predict the phenotype at the individual level.4
Since 2003, Copenhagen University Hospital has offered screening for pathogenic variants in TSC1 and TSC2. From 2017, the screening has been performed using NGS, and since 2018, screening for mosaic pathogenic variants were included. So far, predicted pathogenic variants in TSC1 or TSC2 have been identified in a total of 178 individuals. Since 2015, Aarhus University Hospital, Denmark, has also examined the TSC1 and TSC2 genes using NGS, with an allele frequency of 10% as limit of detection. In the last 3 years, Copenhagen University Hospital has identified pathogenic variants in 12 patients, of which three had pathogenic variants in mosaic state (patients with mosaic TSC). Aarhus University Hospital has identified pathogenic variants in 17 patients with TSC, where two patients had a VAF of 37% and 40%, respectively, and mosaicism could neither be confirmed nor excluded.
Low VAF and mosaicism continue to be hurdles in molecular genetic diagnostics of TSC, but the advent of NGS has improved the potential in diagnostics of patients with mosaicism. The phenotype among patients with mosaic TSC varies considerably, and it is nearly impossible to establish a clear definition of the clinical picture of TSC mosaics; however, a hint may be unilateral angiofibromas and the absence of tubers and subependymal nodules in low-level mosaicism. It is important to discover and diagnose even the mildly affected patients with mosaic TSC, since the descendants can inherit non-mosaic TSC with a more severe phenotype. Therefore, genetic counselling, including thorough clinical as well as a genetic examination, is crucial.
In the case reported here, the patient presented with mild clinical symptoms of TSC in adulthood. When examining the patient, TSC manifestations were found in eyes, skin, and kidneys, and a molecular genetic analysis showed a pathogenic variant in TSC2 with a mosaic level of 10%. It would be interesting to investigate the different affected and non-affected tissues to see if the mosaic frequency is similar in all.





