
Abstract
Common variable immunodeficiency (CVID) is a predominantly antibody deficiency and is one of the most common primary immunodeficiencies in adulthood. Replacement therapy with Ig has significantly reduced infectious complications; however, malignant, autoimmune, and inflammatory diseases are still current major causes of morbidity and mortality. In recent years, interest has increased regarding allergic manifestations that may be associated with primary immunodeficiencies; however, no data are currently available on chronic spontaneous urticaria (CSU). In this report, the authors describe CSU in patients with CVID attending their centre. Three CVID patients were affected by CSU and were unresponsive to antihistamines. Patients were screened for the presence of serum autoreactivity by an autologous serum skin test; only one patient was positive for serum autoreactivity. The serum of this patient was found to induce CD63 upregulation on basophils and degranulation of LAD2 mast cells. All patients were treated with omalizumab therapy at the standard dose of 300 mg every 4 weeks. The patient with autoreactive serum was the best responder to omalizumab therapy, whereas the other two patients experienced urticaria flares related to intercurrent infections. In this article, the authors describe the presence of CSU in patients with CVID for the first time. Although autoimmunity is a feature of CVID, autoreactivity was documented in one patient only, thus showing that CSU in patients with CVID reflects the heterogeneity of this immune defect.
INTRODUCTION
Primary immunodeficiencies (PID) are a large group of rare diseases resulting from genetic errors of the immune system that affect particular immune responses, most of which are usually diagnosed in childhood.1,2 However, some PID may develop in adulthood; for example, predominantly antibody deficiency and diseases such as common variable immunodeficiency (CVID) and IgA deficiency (IgAD).2 CVID is the most common PID in adults and is a heterogeneous group of disorders characterised by the presence of low levels of at least two Ig isotypes (usually IgG and IgA) and a poor response to vaccinations in the absence of other demonstrated causes of hypogammaglobulinaemia.3
The defect predisposes the patient to infections and the development of neoplasia, and it is accompanied by immune dysregulation and autoinflammatory and autoimmune complications.4 Autoimmune diseases occur in approximately 20–30% of patients with CVID, with the most frequent being immune thrombocytopenia and autoimmune cytopenia;5 additional CVID-related systemic and organ-specific autoimmune diseases include systemic lupus erythematosus, pernicious anaemia, antiphospholipid syndrome, multiple sclerosis, Sjögren’s syndrome, psoriasis, thyroiditis, uveitis, vasculitis, and primary biliary cirrhosis, while rheumatologic diseases may be detected in 10–30% of CVID patients.4-7
Among the autoimmune disorders, a peculiar disease is chronic spontaneous urticaria (CSU), which can be considered a disease that overlaps between allergy and autoimmunity.8 CSU is a complex and heterogeneous disease in which autoreactivity is frequently observed. Indeed, one-quarter of patients with CSU display autoantibodies against thyroid autoantigens,9 while 40% show antibodies against IgE or its high affinity receptor (FcεRI) that induces histamine release by mast cells (MC) and basophils.10 An autologous serum skin test (ASST) is used to screen for the presence of autoreactivity in CSU,11 even if its accuracy and reliability are still debated and controversial. An evaluation of the presence of autoantibodies against IgE or FcεRI, as well as the basophil activation test (BAT), can also be performed to confirm serum autoreactivity. However, no correlation between ASST positivity and the presence of autoantibodies has been demonstrated, suggesting that other unidentified factors are responsible for the development of wheals and angioedema.12 Thus, CSU is not a single disease but a pattern of reactions that reflect cutaneous MC degranulation.
Previously, the authors have demonstrated that in vitro evaluation of MC reactivity to CSU serum could be a tool for CSU patient screening.13 Moreover, they have recently described the co-occurrence of CSU in patients with IgAD. In these patients, CSU was associated with autoreactivity, as shown by a positive response to ASST and MC degranulation assays, as well as the presence of other autoimmune disorders.14 Herein, the authors describe three patients with CSU who were also affected by CVID, another PID with predominantly antibody deficiency. In all patients, treatment with omalizumab was effective at controlling or improving symptoms such as wheals and pruritus. The ASST was positive in one patient and MC degranulation and BAT assays were also both positive. In the other two patients, flares of urticaria that were related to concomitant fungal infections were observed.
MATERIAL AND METHODS
Patient Selection
All patients were referred to the Tertiary Centre of Azienda Sanitaria Universitaria Integrata di Udine (ASUIUD), Udine, Italy. A diagnosis of CVID was established in all cases in accordance with the European Society for Immunodeficiencies (ESID) criteria.15 CSU activity was estimated using the Urticaria Activity Score (UAS) according to the European Academy of Allergy and Clinical Immunology (EAACI) guidelines.11 UAS over 7 days (UAS7) was assessed on the basis of symptoms evaluated the week before performing the ASST and again during therapy. The study subjects gave informed consent for participation in this study and publication of the data. At the time of the tests, the patients had stopped taking medication for at least 5 days.
Skin Prick Test
Skin prick tests (SPT) were carried out with commercial extracts as previously described.16 Commercial extracts were provided by Lofarma Allergeni SpA (Milan, Italy) and Stallergenes SpA (Paris, France). A panel of common food allergens (Lofarma Allergeni SpA) was tested. Histamine phosphate and normal saline were used as positive and negative controls, respectively. The response was read 15 minutes after skin puncture and the results were expressed as the mean of the two largest orthogonal diameters in millimetres. A wheal diameter ≥3 mm with erythema compared with the saline control was defined as a positive reaction.
Autologous Serum Skin Test
ASST was performed by an intradermal injection of 0.05 mL of fresh autologous serum and the wheal and flare reaction was recorded after 30 minutes.17
Mast Cell Degranulation and Basophil Activation Test
Evaluation of MC degranulation was performed using the human MC line LAD2 as previously published.13 Briefly, 2×105 LAD2 cells were incubated with 2 µL of serum in Tyrode’s buffer for 30 minutes at 37°C. The reaction was stopped by incubation on ice and the cell suspension was centrifuged. After solubilising the cell pellet with 0.5% Triton X-100 (Sigma-Aldrich, St. Louis, Missouri, USA), the enzymatic activity of the released β-hexosaminidase in the supernatants and cells was evaluated by measuring the amount of 4-p-nitrophenol produced. The extent of degranulation was calculated as the percentage of 4-p-nitrophenol absorbance at 405 nm in the supernatants out of the sum of absorbance in the supernatants and cell pellets solubilised in detergent.
Basophil activation was evaluated using the Fast ImmuneTM CD63/CD123/anti-HLA-DR kit (Becton Dickinson, San Jose, California, USA) following the manufacturer’s indications. Briefly, peripheral heparinised blood was incubated with an equal volume of serum from patients or healthy donors at 37°C for 15 minutes. After red blood cell lysis, basophils were identified as CD63+/CD123+ cells. Sera from three sex and age-matched healthy subjects were used in both BAT and MC degranulation assays as controls. Data were compared by an analysis of variance (ANOVA) test using the post hoc analysis for paired multiple comparisons with Fisher’s least significant difference test.
RESULTS
A computerised search was performed to identify patients with low Ig levels and a diagnosis of urticaria who were referred to the tertiary centre. Eleven patients were identified, five meeting the criteria of CSU and six with a history of acute spontaneous urticaria. Three out of five patients affected by CSU were found to be also affected by CVID.
Patient 1
A 49-year-old female with a history of allergic asthma caused by mites was evaluated for frequent severe exacerbations of asthma in the last 2 years. Asthma exacerbations were associated with airway infections with poor remittance between the acute phases. The diagnostic work-up showed a defect in IgG and IgA levels and bronchiectasis (Table 1). Replacement therapy with intravenous Ig was started with clinical improvement (25 g every 4 weeks). After 1 year, the patient began to complain of CSU that was unresponsive to up-dosed (four-fold) antihistamines (cetirizine). The UAS7 was estimated to be 33 and a SPT for food allergens and ASST were negative. Since asthma was not completely controlled by replacement therapy or high doses of long-acting beta-agonists (formoterol) and inhaled corticosteroids (budesonide), with still frequent use of oral corticosteroids, omalizumab was initiated at a dose of 300 mg every 4 weeks, which improved asthma control and UAS7 (UAS7: 12, data not shown). Although still in therapy with both omalizumab and cetirizine, after 2 years the patient’s symptoms of asthma and urticaria started worsening (UAS7: 38). A SPT for Aspergillus fumigatus was positive. Pulmonary high-resolution CT showed augmented bronchiectasis with a mucus plug and a tree-in-bud pattern, and the patient underwent bronchoscopy. The bronchoalveolar lavage fluid confirmed the presence of Aspergillus. Itraconazole therapy was added to the treatment regimen, resulting in the improvement of asthma and urticaria symptoms. However, as the therapy was interrupted, both asthma and urticaria then worsened. Therefore, vericonazole was prescribed, with a reduction seen in the extent of urticaria (UAS7: 12) and a better control of asthma. Although the patient is in therapy with both omalizumab and antihistamines, wheals and itching are still present. The UAS7 is now estimated to be 12.
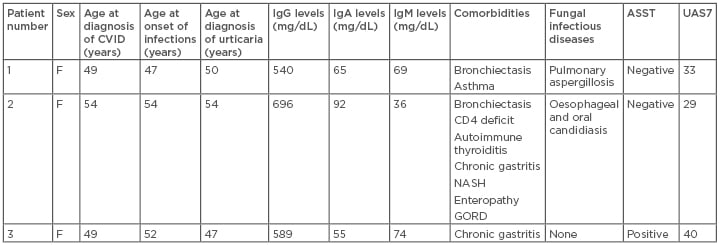
Table 1: Clinical data of Patients 1–3.
Normal values: IgM: 65–210 mg/dL; IgG: 740–1,440 mg/dL; serum IgA: 140–400 mg/dL. ASST was considered positive when the red wheal had a diameter of at least 1.5 mm greater than the control saline solution.17
ASST: autologous serum skin test; CVID: common variable immunodeficiency; F: female; GORD: gastro-oesophageal reflux disease; NASH: nonalcoholic steatohepatitis; UAS7: urticaria activity score over 7 days.
Patient 2
A 47-year-old female was evaluated for the presence of urticaria in the last 3 months (UAS7: 29). Several days before the first evaluation, she underwent endoscopy, which showed the presence of oral and oesophageal candidiasis. Fluconazole therapy was initiated with remittance of the candidiasis. A SPT and ASST were negative, antithyroperoxidase antibodies were present, and low levels of thyroid-stimulating hormone were detected. As shown in Figure 1, IgA and IgG levels were reduced with a concomitant reduction in CD4+ T cells (394 cells/μL). HIV infection was ruled out and the patient was screened for the presence of CVID. Three weeks after the first evaluation, she complained of worsening of urticaria with severe itching and fever. Clinical evaluation, laboratory tests, and chest X-ray were suggestive of the presence of pneumonia, and therapy with amoxicillin/clavulanic acid in association with corticosteroids was prescribed. The diagnostic work-up for CVID showed the presence of bronchiectasis, enteropathy, and autoimmune disorders, such as autoimmune thyroiditis and chronic gastritis with intestinal metaplasia (Table 1). Other disorders not related to CVID were also detected, such as gastro-oesophageal reflux disease and nonalcoholic steatohepatitis. Replacement therapy with intravenous Ig was started (20 g every 4 weeks).
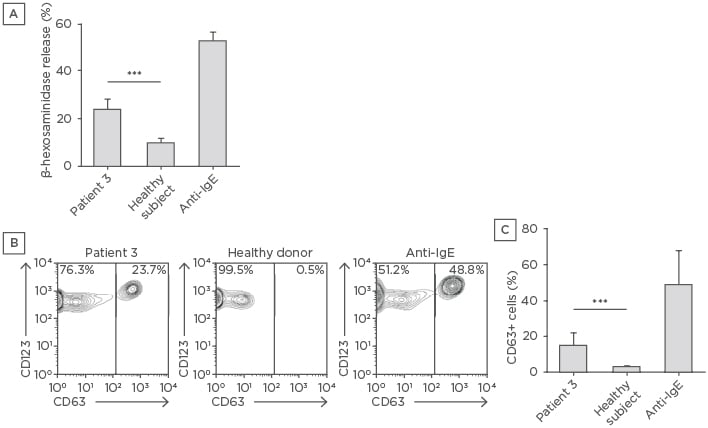
Figure 1: Mast cell degranulation and basophil activation induced by the serum of Patient 3.
A) LAD2 cells were incubated with serum from Patient 3 or a healthy subject. The degranulation response was measured as the amount of released β-hexosaminidase. The data shown are means of three independent experiments. The mean of β-hexosaminidase release obtained with three different healthy sera is shown. B) Peripheral blood was incubated with an equal volume of Patient 3 or healthy donor serum and a fluorescence-activated cell sorting analysis of degranulated basophils identified as CD63+/CD123+ was performed. C) Means ± standard deviations of CD63+/CD123+ cell percentages of two experiments performed in triplicate are shown. The mean of CD63+/CD123+ cell percentages obtained with three healthy sera is shown. Anti-human IgE were used as positive controls in all experiments.
***p<0.001
With resolution of the infections, CSU improved but there still was a significant number of wheals and complaints of pruritus not responsive to high doses of non-sedating second-generation anti-H1 antihistamines (cetirizine). CSU responded to steroids only and, therefore, as soon as replacement therapy was started, the patient underwent therapy with cyclosporine (3.0 mg/kg/day). Cyclosporine was effective at controlling wheals and pruritus (UAS7: 0) but frequent relapses of oral candidiasis occurred and required almost daily fluconazole. On the other hand, the daily dose of cyclosporine could not be reduced without flares of urticaria. Therefore, an off-label therapy with omalizumab at a dose of 300 mg every 4 weeks was started. On omalizumab and replacement therapy, the patient complained of flares of urticaria and oral candidiasis only occasionally. Interestingly, urticaria flares were associated with relapses of oral candidiasis and urticaria completely remitted when candidiasis was treated.
Patient 3
A 47-year-old female developed urticaria after a holiday abroad. She was treated with antihistamines (cetirizine) and steroids with initial response; however, even on therapy with high doses of antihistamines, she had >50 wheals and severe pruritus on a daily basis, which interfered with her sleep (UAS7: 40). She was admitted twice to hospital emergency departments for severe flares of urticaria with angioedema. Initial evaluation ruled out infections due to Salmonella, Shigella, and Campylobacter, and the presence of autoimmune thyroiditis. As shown in Table 1, Ig levels were reduced. Hypogammaglobulinaemia was not related to therapy since low levels of Ig existed before the prescription of corticosteroids. The patient was screened for comorbidities and chronic gastritis was diagnosed. SPT for airborne and food allergens were negative. To characterise the autoimmune nature of the urticaria, patient serum was used to perform an ASST and BAT and to measure MC reactivity; the ASST results were positive. To confirm the autoreactivity of patient serum, the ability to induce a degranulation response of LAD2 cells was analysed; once activated, these cells release β-hexosaminidase.13 As shown in Figure 1A, while sera from healthy controls did not induce a release of β-hexosaminidase, the serum of the patient induced a significant degranulation of LAD2 cells. To further confirm the autoreactivity of patient serum, it was used to stimulate donor basophils in an in vitro heterologous assay (Figure 1B). CD63 expression, detectable on cell membranes only after activation, increased in circulating basophils after incubation with patient serum, while no significant upregulation of CD63 expression was observed using sera from healthy controls (Figure 1C).
Since there was not a clear history of infections, cyclosporine (3.0 mg/kg) was started with slow and progressive improvement. After 1 month, the dose was increased to 4.0 mg/kg and, after 3 months of therapy, the UAS7 was 16. However, there was a slow increase of creatinine blood levels. Within 1 year, CSU was completely controlled (UAS7: 0) and cyclosporine continued to be tapered. Urticaria was completely controlled with a dose of 1.5 mg/kg but, after a few months, the patient decided to stop cyclosporine therapy. After 1 month, wheals and itching started again and could not be controlled by daily 1.5 mg/kg cyclosporine. The daily dose of cyclosporine was progressively increased; however, even at 4.0 mg/kg per day, the UAS7 was 24. Moreover, the patient began to complain of respiratory infections and her creatinine levels rose. At that time, omalizumab became available as a therapeutic option for urticaria and so treatment was shifted from cyclosporine to omalizumab (300 mg every 4 weeks). Within a few days after the first dose of omalizumab, CSU completely disappeared. Although cyclosporine was stopped, the patient still complained of respiratory infections and replacement therapy with subcutaneous Ig (6 g every week) was started. After 6 months of therapy followed by 5 months of therapy with a suspension of 2 months, omalizumab was stopped and urticaria was reported with an estimated UAS7 of 32. Therefore, omalizumab therapy was restarted.
DISCUSSION
CSU is characterised by the development of wheals, angioedema, or both for >6 weeks.11 It is a disorder in which MC play a pivotal role and is thought to be associated with autoimmune mechanisms. Systematic reviews have shown that CSU is strongly linked to autoimmune thyroiditis and that CSU patients have an increased risk of autoimmune disorders, especially adult females and those with a positive family history and a genetic predisposition for autoimmunity.18-20 Autoimmunity is a common feature of PID and autoimmune diseases can be detected in approximately 20–30% of CVID patients.3,4 Many autoimmune disorders are known to be associated with CVID; however, to date, the occurrence of CSU has never been reported in patients with CVID. Here, the authors describe three patients affected by CVID and CSU attending their tertiary centre.
ASST was only positive in one of the patients; as shown in Figure 1, the serum of Patient 3 induced a significant degranulation of the LAD2 MC and a significant increase in CD63 expression on the cell membranes of heterologous basophils. Control sera from healthy donors did not induce either degranulation of LAD2 cells or increased expression of CD63 on basophils. In Patients 2 and 3, omalizumab was effective at controlling urticaria, whereas Patient 1 experienced significant improvements in wheals and pruritus but still reported daily wheals and/or pruritus during omalizumab therapy. Urticaria flares were also observed concomitantly to fungal infections. In Patient 2, flares were strictly related to the reappearance of oral candidiasis, whereas in Patient 1, omalizumab-related improvement was reduced by the onset of aspergillosis; moreover, in Patient 1, urticaria flares were associated with asthma exacerbation. Although an association of urticaria with aspergillosis and fungal infections has been reported,21,22 fungi were not the cause of the CSU in any of the patients since CSU was present even when the patients were in remission for candidiasis and aspergillosis.
There are general concerns regarding treating PID patients with immunosuppressive therapy, but autoimmune and inflammatory diseases are frequently observed in PID and may require different immunosuppressive strategies.23 For example, in CVID patients, cytopenia and some lung and gut diseases require treatment with immunosuppressive agents.3,24 In all CVID patients enrolled in this study, urticaria was resistant even at high doses of non-sedating second-generation anti-H1 antihistamines and patients used systemic steroids for controlling wheals and pruritus. Patients 2 and 3 were initially treated with low doses of cyclosporine since guidelines at the time considered cyclosporine as the next step of therapy, with a moderate level of evidence for efficacy and a moderate safety profile.25 In Patient 2, cyclosporine therapy was started when infections were resolved and when replacement therapy was already established. In this patient, the low level of CD4+ cells was the likely cause of candidiasis and this prompted the authors to shift to a different therapy.
The authors have previously described the co-occurrence of CSU with autoimmune disease and IgAD,12 a PID that shares some clinical features with CVID; these two predominantly antibody deficiencies may occur in members of the same family26 and progression from IgAD to CVID has been reported in several cases.27-30 However, pathogenesis of CSU in CVID seems to be more complex; ASST was positive in only one CVID patient with CSU, whereas serum autoreactivity was a common feature of IgAD patients.14 Moreover, some outbreaks of urticaria were reported in CVID patients that were related to concomitant infections. No IgAD patients with CSU were symptomatic for infection and they were all affected by other autoimmune diseases. The data reported here suggest that CSU pathogenesis in CVID may be more heterogeneous, reflecting the immunological complexity of the underlying disease.
CONCLUSION
Here, the presence of CSU in patients with CVID is described for the first time. A limitation of this study is the small number of enrolled patients because of the rarity of CVID and, more generally, of PID. Therefore, it is speculative to estimate the real co-occurrence of CSU in CVID using a single centre. Although autoimmunity is a feature of CVID, autoreactivity was documented in only one patient. In the other two patients, the authors observed flares of urticaria that were associated with fungal infections. Taken together, these data show that CSU in CVID patients reflects the heterogeneity of immune defects and confirm that CSU is a pattern of MC activation induced by several mechanisms. Further research from different tertiary centres should be used to more effectively estimate the incidence of CSU in CVID patients and to evaluate the possible pathogenetic role of both autoreactivity and underlying infections.





