Meeting Summary
Cardiac amyloidosis is a rare but life-threatening group of disorders caused by the extracellular deposition of misfolded amyloid fibrils in cardiac tissue. Amyloid accumulation leads to cardiomyocyte toxicity, extracellular volume expansion, and ventricular pseudohypertrophy. Two types of amyloid protein are thought to be responsible for most disorders: immunoglobulin light chain, which causes light chain amyloidosis (AL); and transthyretin (TTR), which causes transthyretin amyloidosis (ATTR), of which there are two types: hereditary (hATTR) or wild-type (ATTRwt). Despite increasing clinical recognition of the disease, cardiac amyloidosis remains underdiagnosed. This article explores the epidemiology of AL and ATTR and the noninvasive techniques that help to improve diagnosis of the disorder. Cardiac amyloidosis is associated with mixed phenotype symptoms of polyneuropathy and cardiomyopathy which can lead to multiple misdiagnoses. As a result, patients can wait between 2 and 4 years for a correct diagnosis. Early diagnosis may be aided by recognising red flag symptom clusters. These include family history; neuropathy and sensory involvement; bilateral carpal tunnel syndrome; early autonomic dysfunction and gastrointestinal complaints; heart failure (HF) with preserved ejection fraction (HFpEF; without hypertension); cardiac hypertrophy, arrhythmias, ventricular blocks, right-sided or biventricular HF, or cardiomyopathy; renal abnormalities; and vitreous opacities. Noninvasive imaging techniques have increasingly been used as an alternative to biopsy to diagnose cardiac amyloidosis with the hope of allowing physicians to provide targeted therapy for these patients. Techniques include speckle tracking echocardiography, cardiac MRI, and nuclear scintigraphy, together with biomarkers such as N-terminal pro-brain natriuretic peptide and hepatocyte growth factor (HGF). It is hoped that greater understanding of patients with ATTR may lead to increased awareness of the disorder and improve patient outcomes.
Epidemiological Data1-3
ATTRwt cardiomyopathy is an underdiagnosed but treatable cause of HF; however, the incidence of the clinically relevant disease remains unknown. A study in Umeå, Sweden,1investigated the prevalence of ATTRwt cardiac amyloidosis in >2,200 patients with HF or cardiomyopathy. In the study of 174 patients with HF and interventricular septum >14 mm, approximately 20.0% had ATTRwt amyloidosis, as measured by 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. None of the patients’ blood or urine samples suggested AL, and in the total HF population the prevalence of ATTRwt amyloidosis was 1.1%, giving a prevalence of approximately 1:6,000.1
García-Pavía et al.2 examined demographic and clinical characteristics of patients with ATTRwt using data from the Transthyretin Amyloidosis Outcomes Survey (THAOS).2THAOS is an ongoing international, longitudinal, observational registry that collects data on the natural history of ATTR, and is open to all patients with hATTR or ATTRwt and asymptomatic TTR-variant carriers.3
In the analysis, 758 ATTRwt patients were enrolled in THAOS, most of whom were male (94.6% versus 5.4%), and many patients experienced a significant delay in diagnosis. The mean age at enrolment was 76 years, with a mean age at symptom onset of 68 years, and a mean delay in diagnosis of 3.88 years.2
Almost all patients had either a cardiac (59.8%) or mixed cardiac and neurologic (36.6%) phenotype, highlighting the need for both cardiologic and neurologic assessment of these patients. Neurologic symptoms included sensory (54%), autonomic (34%), and motor neuropathy (29%) (Figure 1). Importantly, the study confirmed that ATTRwt should not be considered an exclusively cardiac disease, but one with a heterogenous clinical spectrum.2
Figure 1: Symptom categories relating to transthyretin amyloidosis reported at enrolment.
Patients with symptomatic transthyretin amyloidosis wild type: n=723. Motor neuropathy includes muscle weakness and walking disability. Sensory neuropathy includes balance abnormality, neuropathic arthropathy, or pain/paraesthesia, numbness, temperature/pain insensitivity, and tingling. Autonomic neuropathy includes dizziness, dry eye, dyshidrosis, palpitations, recurrent urinary tract infections, urinary incontinence, urinary retention, vomiting, constipation, diarrhoea, early satiety, faecal incontinence, nausea, and erectile dysfunction. Other includes carpal tunnel syndrome, endocrine/metabolic disease, eye disease, genitourinary/reproductive disease, inflammatory disease, psychiatric diagnosis, and respiratory disease. Categories are not mutually exclusive. Adapted from García-Pavía et al.2
The Journey to Diagnosis of Hereditary Transthyretin Amyloidosis4
Multiple misdiagnoses can delay the identification of hATTR amyloidosis between 2.0 and 4.3 years.4The difficulty in diagnosis is believed to stem from the multisystemic nature of the disorder.4
Red flag symptom clusters can be used to aid the diagnosis of hATTR amyloidosis. These include:4
- Family history of hATTR symptoms.
- Neuropathy and sensory involvement.
- Bilateral carpal tunnel syndrome.
- Early autonomic dysfunction and gastrointestinal complaints.
- HFpEF (without hypertension).
- Cardiac hypertrophy, arrhythmias, ventricular blocks, right sided or biventricular HF, or cardiomyopathy.
- Renal abnormalities.
- Vitreous opacities.
It is hoped that awareness of these red flag symptom clusters and diagnostic tools could lead to earlier diagnosis of hATTR amyloidosis.4
Imaging Biomarkers: Electrocardiogram and Scintigraphy5-9
Novel imaging techniques are also useful diagnostic tools showcased by Alejandro et al.2who used STE and classical 2D ECG to help differentiate the two subtypes of cardiac amyloidosis: AL and ATTR.5Left and right ventricular apical ratios (LVAR and RVAR) were compared among 78 patients with cardiac amyloidosis (47 with AL and 31 with ATTR) and 24 healthy controls. Both LVAR and RVAR were elevated in AL compared with ATTR.
Cut-off values of LVAR >0.96 and RVAR >0.80 showed high accuracy in differentiating the two subtypes. The study authors concluded that RVAR is an easy and accessible tool for most echo laboratories to help differentiate AL and ATTR.5
These findings were supported by Zhang et al.6who used the apical sparing ratio to detect differences among 179 patients with suspected AL and ATTR cardiac amyloidosis (Figure 2). AL versus TTR was determined by mass spectrometry, if available, and TTR was assigned if biopsy, serum protein, and/or free light chain testing was negative for AL. Patients with negative endomyocardial biopsy or autopsy were included as controls. They reported that a lower ratio cut-off (0.81) than previously reported had optimal diagnostic sensitivity (72%) and specificity (64%).6
Figure 2: Apical sparing ratio was higher in transthyretin than light chain amyloidosis.
Routine longitudinal strain imaging. The apical sparing ratio was calculated as the average of apical segments/average of mid + average of basal segments. ANT: anterior; ANT_SEPT: anteroseptal; INF: inferior; LAT: lateral; POST: posterior; SEPT: septal. Adapted from Zhang et al.6
How does ATTR affect patient outcomes over the long term? Recent studies have shown a relatively high prevalence of ATTR in patients with aortic stenosis, the most common valvular heart disease in the Western world. A study from Israel by Zikry et al.7sought to examine the effects of ATTR in 86 elderly patients (mean age: 81 years) who underwent surgical aortic valve replacement or transcatheter aortic valve replacement therapy.7
Using nuclear cardiac imaging with 99mtechnetium-pyrophosphate (99mTc-PYP), patients were assessed for ATTR over three timepoints: before aortic valve replacement therapy, 1 month after, and 2 years after the procedure. Patients who were positive for ATTR (14%) had a more severe clinical presentation and more advanced signs of HF than patients without ATTR. Before the intervention, the hospitalisation due to HF rate was 3.26-times higher in patients with ATTR compared to those without ATTR (p=0.01). After the intervention, diastolic function remained more severely affected in the ATTR-positive group at follow-up than in the ATTR-negative group (p=0.05). Furthermore, hospitalisation rates after intervention of HF were 2.84-times higher in patients with ATTR compared to those without (p=0.02). The findings suggested that older patients with amyloidosis have a more severe prognosis following aortic valve replacement therapy than those without amyloidosis.7
Diagnosis of ATTR in patients undergoing valvular intervention is of importance as it may affect their clinical outcomes and the need for pacemakers. In an accompanying presentation, Zikry et al.8used 99mTc-PYP scanning to examine ATTR among 86 older patients (mean age: 78 years) undergoing transcatheter aortic valve implantation (TAVI). They found that 12 (14%) patients were positive for ATTR and that conduction abnormalities were significantly more prevalent in these patients. In addition, 42% of ATTR-positive patients underwent periprocedural permanent pacemaker implantation compared to 28% implantations in the ATTR-negative group (p=0.043). There was also a significantly higher rate of new left bundle branch block development in the ATTR-positive group compared to the ATTR-negative group (39.1% versus 10.9%; p=0.03). They concluded that patients undergoing TAVI should be observed carefully for signs of conduction disorders and possible pacemaker insertion.8
ATTR is an underdiagnosed disease presenting with HFpEF symptomatology; therefore, Fukuzawa et al.9used 99mTc-DPD/MDP/HMDP bone scan scintigraphy to determine the prevalence of ATTR among 62 patients aged ≥60 years with HFpEF (left ventricular ejection fraction ≥50%). Relatively intense myocardial uptake of 99mTc-PYP was found in 7 (11%) patients, with diffuse deposition in both right and left ventricles. Patients with amyloid deposition were also older and had lower systolic blood pressure and left ventricular ejection fraction. It was suggested that the findings may allow the possibility of targeted therapy for these patients.9
Biological Markers and Echocardiogram Abnormalities in Cardiac Amyloidosis
Cardiac amyloidosis poses significant diagnostic and therapeutic challenges. The disorder is particularly difficult to detect early as patients can become symptomatic before any signs of cardiac amyloidosis involvement is suspected. Classical tests such as ECG pseudo-infarct and concentric hypertrophy, as well as delayed gadolinium enhancement on MRI, may lack diagnostic specificity and sensitivity if misinterpreted. Once cardiac amyloidosis involvement has been established, it is associated with poor survival.10
As multiple new therapies for AL and ATTR amyloidosis emerge, there is an increased need to identify relevant serum biomarkers that may improve prognostic and staging systems; however, there is currently a lack of international guidelines on the use of biomarkers in these patients.10
A potential biomarker for AL and ATTR cardiac amyloidosis is HGF. HGF is a potent angiogenic growth factor that is thought to be released during amyloid deposition in the myocardium. In their study, Zhang et al.10enrolled 102 patients with suspected cardiac amyloidosis, and levels of HGF, N-terminal pro-brain natriuretic peptide (NT-proBNP), troponin-T, and estimated glomerular filtration rate were measured. Cardiac involvement was established via endomyocardial biopsy; if this was not available, transthoracic echocardiography, ECG, and cardiac MRI were used. Patients were then followed for all-cause mortality, cardiac transplant, and left ventricular assist device implantation. Of the 102 patients enrolled, 72 had known cardiac involvement. Patients were stratified using biomarker cut-off levels established from previously published models (Table 1).10
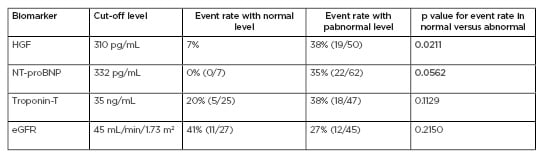
Table 1: Elevated hepatocyte growth factor and N-terminal pro-brain natriuretic peptide and clinical outcomes in amyloidosis.
eGFR: estimated glomerular filtration rate; HGF: hepatocyte growth factor; NT-proBNP: N-terminal pro-brain natriuretic peptide. Adapted from Zhang et al.10
Results found that elevated HGF was associated with worse clinical outcomes resulting in all-cause mortality, left ventricular assist device implantation, and cardiac transplant. Kaplan–Meier analysis of survival stratified by HGF level revealed rapid divergence in survival time for HGF levels >310 pg/mL. Although NT-proNBP also predicted similar outcomes, they were not specific to cardiac amyloidosis, therefore, future studies will compare the diagnostic and prognostic specificities of HGF and NT-proNBP in patients with cardiac amyloidosis.10
Cardiac troponins are also chronically elevated in cardiac amyloidosis. Castiglione et al.11assessed the prognostic value of high-sensitivity cardiac troponin-T (hs-cTnT) and NT-proBNP in 230 patients with suspected cardiac amyloidosis. Cardiac amyloidosis was confirmed in 86 (37%) patients, 29% of whom had AL and 71% ATTR. Further results found that patients with cardiac amyloidosis had higher plasma levels of NT-proBNP (5,507 ng/L [2,348–10,326] versus 1,332 [392–3,752]; p<0.001) and hs-cTnT (65 ng/L [48–114] versus 35 [21–52]; p<0.001) than those with suspected, but not confirmed, cardiac amyloidosis. The combination of the two biomarkers improved discrimination over NT-proBNP alone (p=0.011), but not over hs-cTnT (p=0.470). An NT-proBNP level <600 ng/L or an hs-cTnT level <17 ng/L was optimal for ruling out cardiac amyloidosis (95% negative predictive value for both).11
How common are ECG abnormalities in patients with cardiac amyloidosis? In their retrospective, observational study of 251 patients with cardiac amyloidosis, Martone et al.12found that three-quarters of the patients had ECG abnormalities (82% in ATTR versus 72% in AL; p=0.06). Rhythm disturbances were more prevalent in ATTR as evidenced by a higher burden of IV conduction delays (43% versus 21%; p<0.001) and a higher prevalence of pacemakers (24 versus 1 patient) compared with AL. After adjusting for age and sex imbalance, ATTR was independently associated with a higher prevalence of atrial fibrillation and atrioventricular conduction delays compared to AL (adjusted odds ratio: 4; 95% confidence interval: 1.4–11.2; p=0.008 and adjusted odds ratio: 6.2; 95% confidence interval: 2.6–14.9; p<0.001, respectively). By contrast, AL was associated more with low-voltage QRS.12
Conclusion
In summary, cardiac amyloidosis is a rare, life-threatening disease that can be challenging for physicians to diagnose due to multisystem involvement.4Highly specific, noninvasive cardiac tests, such as nuclear scintigraphy, have helped improve diagnosis of the disorder and offer an alternative to cardiac biopsy.4Plasma biomarkers, including NT-proBNP and HGF, also offer potential as diagnostic and prognostic indicators.10These techniques may lead to a better understanding of the disorder with the aim of improving patient outcomes.






