Meeting Summary
The meeting’s objectives were to review the principles in diagnosing Fabry disease according to the European Society of Cardiology (ESC) guidelines on hypertrophic cardiomyopathy (HCM); to discuss the practical challenges in diagnosing Fabry disease in clinical practice; to investigate the long-term benefit of enzyme replacement therapy (ERT) for patients with Fabry disease; and to identify key patient populations with Fabry disease at risk of misdiagnosis.
Prof Aleš Linhart opened the symposium by highlighting that a significant number of cardiologists are not aware of Fabry disease and that the average time to diagnosis is >10 years.1 The need for treatment of rare cardiomyopathies was also discussed. Prof Perry Elliott reviewed the ESC guidelines on diagnosis and management of HCM, and how they apply to Fabry disease. Prof Linhart then outlined how these guidelines can practically be applied, using case studies to illustrate the challenges in accurately identifying patients with a potential diagnosis of Fabry disease. Prof Linhart then demonstrated the long-term benefits of ERT for patients diagnosed with Fabry disease observed in Mainz, Germany, on behalf of Prof Christoph Kampmann, while Assoc Prof Jean-Claude Lubanda highlighted key patient populations with an increased prevalence of Fabry disease who should be targeted for screening to improve therapy and clinical outcomes.
Introduction
Professor Aleš Linhart
Diagnosing the underlying disorder in a patient presenting with a cardiomyopathy, defined as a structurally and functionally abnormal heart muscle not caused by abnormal loading conditions or coronary disease, can be difficult.2 Cardiomyopathies can be isolated or associated with a variety of extracardiac symptoms and the pathology may have a genetic or non-genetic origin.2 Therefore, the ESC has issued guidelines to assist healthcare professionals in accurately diagnosing the underlying aetiology of hypertrophic cardiomyopathies.
Using ESC Guidelines in Clinical Practice to Identify Patients with Fabry Disease
Professor Perry Elliott
HCM is defined by an increased left ventricular (LV) wall thickness that cannot be explained by abnormal loading conditions.2 In adults, HCM is defined by a LV wall thickness of ≥15 mm in one or more myocardial segments, whereas in children, HCM is defined by a LV wall thickness of more than two standard deviations above the age and size-predicted means. It is recognised that a variety of genetic and non-genetic diseases may underpin this clinical presentation, which influences both the treatment modality used and the prognosis, and in 40–60% of cases HCM is related to a sarcomeric protein gene mutation (Figure 1).
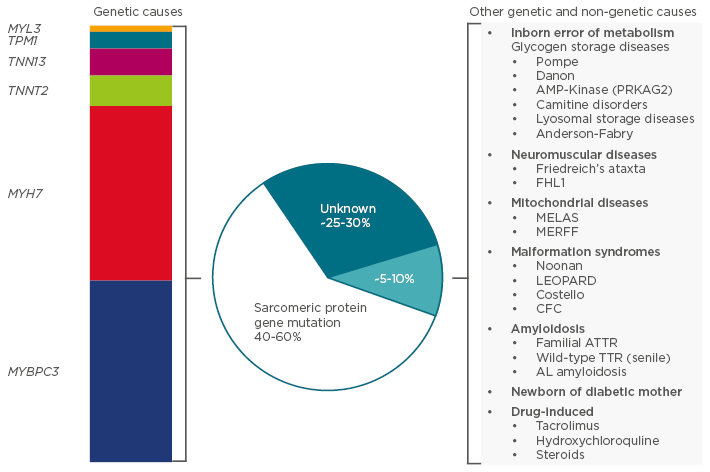
Figure 1: Aetiology of hypertrophic cardiomyopathy.
MELAS: mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke; MERFF: myoclonic epilepsy and ragged red fibre disease; LEOPARD: lentigines, electrocardiographic conduction defects, ocular hypertelorism, pulmonary stenosis, abnormalities of the genitals, retarded growth, deafness; CFC: cardiofaciocutaneous syndrome; ATTR: transthyretin-related amyloidosis; TTR: transthyretin; AL: amyloid light-chain.
Adapted from the 2014 Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the ESC Guidelines.2
Consequently, the underlying cause of increased LV wall thickness in the absence of loading abnormalities should be clinically investigated. This is of particular importance as numerous genetic disorders are associated with an increased LV wall thickness, including Fabry disease and amyloidosis, which may present as HCM phenocopies (diseases with similar clinical phenotypes).2 Although HCM phenocopies are rare, their exclusion or identification is critical for disease management, as delays in diagnosis can worsen prognosis.3
Several cardiac investigative techniques, such as electrocardiogram (ECG), echocardiogram, and cardiac magnetic resonance imaging (MRI), are able to identify diagnostic ‘red flags’, which can assist in making a diagnosis of Fabry disease when assessed in conjunction with age, clinical history, and family history. Some ECG features can be indicative of specific diseases and useful for selecting further tests to help obtain a definitive diagnosis. For example, a short PR interval with pre-excitation in a young patient can be a normal variant, whereas in an older patient (>30 years) with a short PR interval, no pre-excitation, and with concentric left ventricular hypertrophy (LVH), could indicate Fabry disease.2,4 Images obtained from an echocardiogram can also provide further evidence towards a diagnosis; for example, if an older patient (>70 years) presents with concentric hypertrophy, where every segment is the same thickness, it provides evidence of mitochondrial disease or amyloidosis. Cardiac MRI of the interventricular septum is also advantageous in facilitating a differential diagnosis, where a reduction in T1 signal in a patient with concentric LVH is a feature almost exclusive to Fabry disease.5
Recent years have also seen the adaptation of old diagnostic tests for new applications. Use of the bone tracer technique 99m-technetium-labelled 3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy has been demonstrated to be a highly sensitive and specific test for accurately diagnosing patients with transthyretin-related amyloidosis.6 When used in a prospective cross-sectional study in Spain, it was found that 13% of patients with heart failure with a preserved ejection fraction have amyloidosis, suggesting that the prevalence of amyloidosis may be underestimated.7
The ESC guidelines also recommend the use of simple blood tests in identifying some causes of HCM. Examples include the measurement of alpha-galactosidase A in men, to confirm a diagnosis of Fabry disease, and serum creatine phosphokinase levels, which are raised in a variety of metabolic and neuromuscular disorders.2,8 Genetic screening may be warranted for the relatives of patients fulfilling the diagnostic criteria for HCM,2 however a possible barrier to the adoption of genetic testing is the limited knowledge amongst cardiologists regarding when to initiate a genetic test and how to interpret the results.
In conclusion, a multidisciplinary approach should be used when diagnosing the underlying cause of HCM, as many conventional cardiac tests, when used independently, are insufficient for generating a definitive diagnosis. In the case of Fabry disease, clinicians need to be aware of diagnostic red flags, such as a short PR interval on an ECG and a reduction in the T1 signal on a cardiac MRI, and consider these in conjunction with laboratory tests, symptoms, and family history, prior to making a diagnosis.
Guess Who: Can You Spot the Patient with Fabry Disease? Putting Guidelines into Practice
Professor Aleš Linhart
Patients generally present to their doctor with symptoms and a personal history, not details of an underlying genetic disorder. Patients with Fabry disease may not present with cardiovascular symptoms, but anyone referred to a cardiologist is likely to undergo basic tests, such as an ECG, echocardiography, and cardiac MRI.9 A high proportion of patients with Fabry disease present with HCM, particularly concentric diffuse hypertrophy.10-12 In particular, posterior or posterolateral scars that do not appear to be a result of ischaemia when associated with HCM may be a marker of Fabry disease.13 Alternatively, patients with Fabry disease may present with a mitral valve prolapse.10
However, HCM is a phenotype that is shared by many genotypes. For example, in the instances where Fabry disease patients present with asymmetric septal hypertrophy, it may be difficult to distinguish the structural differences in the myocardium from those observed in patients with sarcomeric HCM.10 Therefore, it is important for cardiologists to investigate issues involving any other organs as this can provide insight into the underlying disorder.
As a syndrome, classical multi-organ disease may first be observed in patients with Fabry disease, including kidney ailments and strokes starting at 20 years of age.12 In particular, patients presenting with a late-onset variant of Fabry disease will present largely with cardiomyopathy, possibly alongside cornea verticillata and microalbuminuria or proteinuria.12 However, in a phenotype that suggests kidney damage could be symptomatic of many different cardiomyopathies, endomyocardial biopsies and genetic analysis may be necessary to confirm a diagnosis of Fabry disease. A renal biopsy showing typical lesions within the glomerulus and zebra bodies on electron microscopy, resulting from globotriaosylceramide being stored within the lysosomes, may also be indicative of Fabry disease.14
As Fabry disease is an X chromosome-linked disease that presents as a syndrome that can affect almost any system in the body, investigating family history may also be useful in obtaining a diagnosis, as a clear-cut pattern of inheritance could provide an indication of the underlying disease.15 However, the X-linked inheritance pattern of Fabry disease can also make a diagnosis guided by family history difficult, because heterozygous females can be oligosymptomatic, if not asymptomatic, meaning that familial inheritance may not be immediately apparent.16
Therefore, identifying and correctly diagnosing patients with Fabry disease can be difficult and somewhat of a puzzle. While typical patients are described in the literature as having diffuse HCM with extracardiac manifestation, proteinuria, renal insufficiency, nephropathy, and/or skin lesions, not all patients will present with this phenotype.12 Therefore, Fabry disease should be considered for all patients with unexplained LVH. While it is important to diagnose Fabry disease early when ERT treatment is most effective, an accurate diagnosis in patients with later-stage disease is also important because it may allow renal function to be stabilised, preventing further nephropathy progression.3,12
Ten Years of Enzyme Replacement Therapy: Long-Term Treatment Outcomes in Fabry Disease
Professor Aleš Linhart on behalf of Professor Christoph Kampmann
ERT for patients with Fabry disease became widely available approximately 15 years ago.17 Since then, there has been increasing interest in long-term outcomes for patients diagnosed with Fabry disease. Cardiac alterations that manifest as dyspnoea, angina, fatigue, and palpitations are one of the predominant features of Fabry disease.18 These symptoms are often a result of progressive HCM,18 so the long-term effect of ERT on progressive HCM is of interest.
A single-centre, retrospective study aimed at evaluating the long-term effectiveness of agalsidase alfa on the progression of cardiomyopathy in patients with Fabry disease, reviewed the records of 45 patients (21 male, 24 female) with confirmed Fabry disease who had been treated with agalsidase alfa 0.2 mg/kg intravenously every other week over a median of 10.8 years.19 Results from the study demonstrated that long-term treatment with agalsidase alfa improved or stabilised symptoms of heart failure, with 13 out of 14 patients with New York Heart Association (NYHA) Class II or III heart failure improving by at least one functional class.19 Only one patient in the study (baseline: NYHA Class I) exhibited deterioration of heart failure symptoms.19 Angina symptoms, measured using the Canadian Cardiovascular Society (CCS) scoring system, also improved (15/42; 36%) or stabilised (26/42; 62%) in most patients.19 Furthermore, CCS angina scores for all patients with a baseline score of II or above (n=11) were reduced to score I or lower following ERT.19
When discussing the impact of ERT on LVH, it is important to place any results in the context of the progressive nature of cardiac alterations over the course of the disease.18 After 10 years of ERT, symptoms of cardiomyopathy were stable or improved, and progression was generally attenuated in both males and females.19 In patients administered agalsidase alfa therapy, LVH progression also stabilised with no significant change in left ventricular mass index (LVMI) in male patients with a baseline LVMI <50 g/m2.7 and in all females (Figure 2).19 In males with a baseline LVMI ≥50 g/m2.7 (n=15), LVMI was significantly reduced after 10 years of agalsidase alfa therapy (p=0.0061) (Figure 2).19 Cardiac and renal function, measured by LV ejection fraction and estimated glomerular filtration rate (eGFR), respectively, also remained stable after 10 years of treatment.19
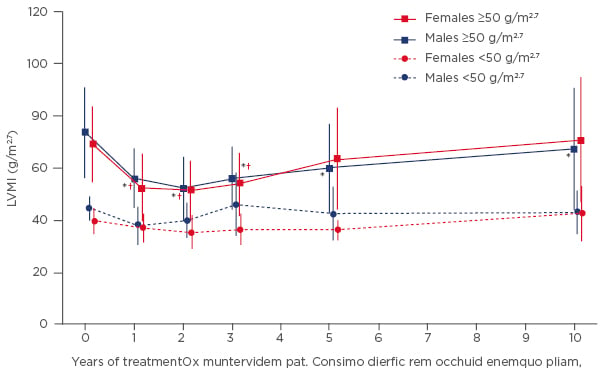
Figure 2: Mean left ventricular mass index (±SD) during 10 years of agalsidase alfa therapy stratified by gender and baseline left ventricular mass index.
*p<0.05 versus baseline for males with LVMI ≥50 g/m2.7; †p<0.05 versus baseline for females with LVMI ≥50 g/m2.7.
LVMI: left ventricular mass index; SD: standard deviation.
Adapted from Kampmann 2015.19
Notably, whilst similar studies have been performed,20,21 few have managed to account for multiple cardiac and renal parameters, such as heart failure and angina status, LVH, eGFR, and proteinuria, over such a long duration of treatment. However, these outcomes are largely supported by a recent retrospective analysis of the Fabry Outcome Survey (FOS), which assessed cardiac and renal outcomes in patients receiving ERT over 10 years.22
FOS is an international registry which includes patients with Fabry disease who are receiving, or are candidates for, ERT with agalsidase alfa.23 The recent analysis incorporated data from three cohorts:22
- Patients (n=382) who received agalsidase alfa therapy for a minimum of 10 years
- A renal cohort (n=105) taken from the treated cohort, with at least three available eGFR measurements
- A cardiac cohort (n=50) who had at least three available LVMI measurements
Analysis of the cardiac cohort demonstrated that over 10 years of agalsidase alfa therapy, LVMI stabilised among patients with no LVH at baseline.22 In patients with LVH at baseline, small increases in LVMI were evident over time.22 In addition, renal function remained relatively stable in females and demonstrated non-significant decreases in males.22
Long-term evidence from a single-centre study and the FOS analysis demonstrated that treatment with agalsidase alfa over 10 years stabilised or improved cardiac symptoms and cardiac structure.19,22 In addition, renal function is generally preserved and cardiac function stabilised over time.19,22 Although limited by small numbers because of the rarity of Fabry disease, these findings are supported by similar studies of shorter duration, and suggest that agalsidase alfa offers long-term benefits for patients with Fabry disease-associated cardiomyopathy.19
What is the Value in Screening Programmes for Rare Diseases?
Associate Professor Jean-Claude Lubanda
Diagnosing Fabry disease can be challenging for clinicians because patients can present with symptoms that are synonymous with several conditions, including pain, paraesthesia, hypohidrosis and hyperhidrosis, or angiokeratomas.12 Additionally, physical examinations and imaging tests are extremely relevant for correctly identifying cases of Fabry disease, particularly in patients who present with atypical symptoms.24
Fabry disease is caused by a deficiency in the a-galactosidase A (a-GAL A) enzyme encoded by the GLA gene.25,26 Assays of α-GAL A activity are not considered to be reliable enough to form a diagnosis, especially as there are differences in activity between males and females. Therefore, genetic analysis of the GLA gene is considered to be a more appropriate diagnostic method and is the only reliable test for Fabry disease females.27 Nevertheless, measuring plasma globotriaosylsphingosine (lyso-Gb3) levels as part of a dried blood spot test may be useful because it is an important biomarker for the pathogenesis of Fabry disease.28,29 However, genetic confirmation in females is still required for a final diagnosis.28
Given the numerous tests that can be used to screen for Fabry disease, the question of which specific population should be subjected to extensive testing still remains. Historically, family screening has been the most commonly used method for diagnosing Fabry disease. While screening newborns may have benefits, the low prevalence of 0.03% makes this challenging.25 Therefore, screening methods tend to focus on evaluating the prevalence of Fabry disease in high-risk populations.
Cardiologists are specifically interested in patients with LVH, a distinguishing feature of Fabry disease. For example, screening of patients with LVH and low plasma a-GAL A activity, but no other symptoms of Fabry disease, indicated that approximately 3% of patients had Fabry disease-related mutations.30 A similar prevalence of 4% was reported following systematic screening for undiagnosed Fabry disease in patients with LVH by another group.31 Patients with end-stage kidney disease are another target population for plasma α-GAL A screening given severe kidney disease is also a common symptom of Fabry disease. Within this population, Fabry disease confirmed by gene mutation has a reported prevalence of 0.7%,32 and 0.2% amongst patients undergoing haemodialysis assessed using dried blood spot screening.33 This highlights how patients with Fabry disease, whose symptoms are classified into broad categories (e.g. HCM), or who suffer comorbid diseases, can be misdiagnosed and given ineffective treatment, which can negatively affect their prognosis. Additionally, in the case of Fabry disease, a positive diagnosis and subsequent screening of other members of the family can facilitate early treatment, improving an at-risk patient’s prognosis.15 Other populations that could potentially benefit from screening for Fabry disease include patients with other unexplained symptoms, such as cryptogenic stroke. Surprisingly, 4.9% of males and 2.4% of females who have suffered a cryptogenic stroke were found to have GLA mutations after being screened for a-GAL A activity.34
Overall, it has been demonstrated that screening for Fabry disease, despite its rarity, is important. There is now evidence to suggest that the prevalence of Fabry disease has been historically underreported because of the variability in symptom presentation. Patients with LVH, end-stage kidney disease, or cryptogenic stroke have a relatively high prevalence of Fabry disease that may justify routine screening, ultimately leading to more effective treatment for these patients. This is especially important in atypical cases where patients may not present with any other symptoms of the disease. Additionally, a positive diagnosis for Fabry disease following screening may allow a patient’s family members to also be screened and to receive early treatment, if necessary, improving their prognosis.
Question and Answer Session
Q: Does every patient with HCM need to be tested for Fabry disease?
Prof Linhart suggested that a test for Fabry disease should be included in the panel of tests used for all patients with HCM, especially because there can be overlap between the clinical features of Fabry disease and sarcomeric gene mutations. Also, if sarcomeric gene mutations have been excluded, this increases the probability that a patient with HCM has Fabry disease.
Q: Should an 18-year-old patient with LVH be tested for Fabry disease?
Prof Linhart replied that registry data indicate that significant LVH, that meets the criteria for HCM, is unlikely to be present in Fabry disease affected males aged <25, or females aged <30. However, Fabry disease should not be discounted in patients who have reached these ages and have mild LVH.
Q: Should different screening tests for Fabry disease be used for men and women?
Assoc Prof Lubanda outlined that enzyme testing in women is problematic, because enzyme levels may appear normal in female patients with Fabry disease. Therefore genetic testing is always recommended, especially in women, but enzyme activity testing offers a useful screening method for men because low activity is a reliable sign of the disease. Genetic confirmation of a pathogenic mutation in the GLA gene is mandatory for a final diagnosis.
Q: When should endomyocardial biopsy be used for diagnosing Fabry disease?
Prof Linhart indicated that different mutations that can lead to Fabry disease can have differential effects on the patient. Therefore, looking for lysosomal lyso-Gb3 storage in endomyocardial biopsies following an enzymatic or genetic diagnosis of Fabry disease is useful when confirming the heart is impacted by the disease.