
Abstract
Inflammation and thrombosis are interrelated processes that are important in the pathogenesis of atherothrombosis. Inflammation is important in both the early and late stages of atherosclerosis, and it involves elements of immune system activation. Low density lipoprotein (LDL) is an important initiator but is not the only one. LDL enters the cell membrane, is modified, and sets into motion a series of events that stimulate the ingress of specific proinflammatory mononuclear cells through the vessel wall. These cells imbibe lipids and form foam cells. Proinflammatory mediators secreted by these cells can eventually lead to intimal thickening and lipid accumulation, forming atherosclerotic plaques. A complex interplay between inflammation, platelet function, and hypercoagulability is a major contributor to the progression from stable to unstable plaque and an acute coronary event. In the later stages of atherosclerosis, inflammatory cells can destabilise certain lipid-rich lesions contributing to symptomatic coronary thrombosis. Thus, thrombosis is the final common pathway for most atherosclerotic complications. Thrombi may also contribute to the asymptomatic rapid progression of atherosclerotic lesions. While antithrombotic agents are important in the treatment of acute coronary syndromes, as well as preventive therapy in high-risk primary prevention and in secondary prevention, the role of specific anti-inflammatory agents is not currently established. If such therapies are to become routine, these anti-inflammatory drugs must significantly reduce events while not adversely affecting a patient’s natural immunity to an extent that erases any potential benefit. This article reviews these two processes with an emphasis on coronary atherosclerosis and its sequelae.
INTRODUCTION
The importance of inflammation and thrombosis in the pathophysiology of atherosclerosis was debated in the 19th century by two prominent pathologists, Virchow and Rokitansky, who had opposing views. In the 20th century, the role of thrombosis in the pathogenesis of acute myocardial infarction remained unsettled until the 1970s.1 Studies over the last 40 or more years have improved the understanding of these two processes and explained the role of inflammation in the initiation and progression of atherosclerosis. There is a close interrelationship between inflammation, which is a driver of the atherosclerotic process, and most thrombotic lesions, the final pathway that leads to many of the complications triggered by inflammation. This article reviews some of these data and considers the clinical importance of antithrombotic and anti-inflammatory agents in mitigating these processes. The emphasis of this review will be on coronary atherosclerosis and thrombosis.
INFLAMMATION AND ATHEROSCLEROSIS
Dr Russell Ross, a pioneer in the field of atherosclerosis pathogenesis in the second half of the 20th century, considered atherosclerosis to be a chronic inflammatory disease of the intima.2 Atherosclerosis principally affects large and medium-sized elastic and muscular arteries, and it may lead to ischaemic complications in the muscles served by these arteries. It is well known that early atherosclerotic lesions can be found in adolescents and young adults decades prior to any clinical sequelae. Pathologic studies in children at autopsy, autopsy studies in young adults during war time, and intravascular ultrasound studies of the coronary arteries in adolescent and young adult donor hearts soon after transplant attest to the common presence of fatty streaks or intimal thickening, particularly if risk factors are present.3-5 The next section will consider a brief description of the role of inflammation in early atherosclerosis. For a more detailed discussion, the reader is referred to the report by Libby et al.6
The inflammatory response in atherosclerosis involves elements of both the innate (e.g., the most prominent cells are certain proinflammatory monocytes) and the adaptive (e.g., T lymphocyte subtypes) arms of the immune system. Considerable evidence supports the importance of oxidised phospholipids from modified low-density lipoproteins (LDL) as an important, but not the only, initiator for the ingress of monocytes into the intima by eliciting the expression of adhesion molecules and chemoattractant molecules, including chemokines, such as monocyte chemoattractant protein-1.7 Pathologic studies of the coronary arteries in young adults dying from noncardiac causes indicate that lipid accumulation in the intima precedes intimal inflammation.8 Once in the intima, monocyte-derived macrophages imbibe oxidised lipids to form foam cells. These cells secrete chemoattractant chemokines that encourage the migration of smooth muscle cells from the media into the intima. An intimal accumulation of smooth muscle cells in a proteoglycan-rich matrix (secreted by the smooth muscle cell) with extracellular lipid forms a progressive lesion termed pathologic intimal thickening. It is generally accepted that advanced atherosclerotic lesions responsible for the development of acute coronary events develop from intimal masses and not necessarily from fatty streak, which often regress.9
A discussion of coronary atherosclerosis must also consider the role of altered haemodynamics and flow patterns in the development of focal atherosclerotic lesions within the coronary arteries. Local haemodynamic forces and, in particular, low endothelial shear stress, significantly contribute to early lesion development and progression of atherosclerosis.10 Low shear stress modulates endothelial gene expression, inducing an atherogenic phenotype to the endothelium. Low shear stress also promotes inflammation by activating nuclear factor κB and by reducing the bioavailability of endothelial nitric oxide.11
As lesions progress, a fibrous cap forms, composed of smooth muscle cells with an extracellular matrix of collagen and elastin infiltrated by inflammatory cells (macrophages and T lymphocytes to a varying degree). Underlying the cap, there may be a mass of lipid composed of extracellular lipid and necrotic cells derived, in part, from the apoptosis of macrophage-derived foam cells; these come together to form the necrotic core. The lipid can be found as either cholesterol esters, free cholesterol, or monohydrate cholesterol crystals.
Vasa vasorum grow into the intima from the adventitia as plaques enlarge to provide nourishment to the thickened vessel wall. Intraplaque haemorrhage from these thin-walled vessels with faulty endothelial cell junctions is a potential source of additional lipid and inflammatory cells allowing for asymptomatic, accelerated growth of the plaque.12 Not all plaques contain an abundance of lipids. Other plaques are fibro calcific, with little or no lipid core, and they are potentially not as dangerous as the lipid-rich plaques, particularly those with a thin fibrous cap that are classified as thin-capped fibro atheromas (TCFA).
INFLAMMATORY MARKERS AND CORONARY ARTERY DISEASE
There is abundant literature concerning the role of biomarkers in atherosclerosis. High sensitivity C-reactive protein (hsCRP) is the most widely studied. It is a sensitive but non-specific marker of inflammation that, when elevated to >3 mg/L, has been shown to be associated with a significant increase in coronary events compared to a normal value.13 HsCRP has been added to conventional risk scores in the Reynolds Risk Model and improved prediction above the Framingham Risk Score.14 In the JUPITER Study, patients with an elevated value ≥2 mg/L and LDL levels <130 mg/dL were randomised to rosuvastatin 20 mg versus standard care. These patients had a 44% reduction in a composite endpoint, including myocardial infarction, stroke, and cardiovascular death.15 More recently, in an analysis from the FOURIER trial,16 LDL cholesterol reduction with the PCSK-9 inhibitor, evolocumab, reduced cardiovascular events across hsCRP strata in patients with clinically stable cardiovascular disease with greater absolute risk reductions in patients with higher baseline hsCRP. Furthermore, in patients with LDL levels as low as <20 mg/d L on therapy, 3-year event rates were higher in those with an elevated hsC-RP compared to those with a level <1 mg/L.16
The CANTOS trial assessed the role of canakinumab, a monoclonal antibody targeting IL-1β in patients with a prior myocardial infarction and an elevated hsCRP.17 This was a test of the inflammatory hypothesis in atherosclerosis, because the drug does not alter lipid levels. At a median follow up of 37 months, the drug reduced the primary endpoint of myocardial infarction, stroke, or cardiovascular death by a small, but significant, amount compared to placebo. However, the residual event rate was high and canakinumab was associated with a significant increase in fatal infections versus placebo.
FROM LESION PROGRESSION TO SYMPTOMATIC THROMBOSIS
Asymptomatic progression of atherosclerosis can be linear or non-linear, the latter caused by processes that acutely (or subacutely) accelerate plaque burden. This is generally related to two processes. The first has already been alluded to: intraplaque haemorrhage from rupture of the vasa vasorum. The other process is asymptomatic thrombosis. Pathologic studies in patients that died as a result of sudden cardiac death frequently show multiple prior episodes of asymptomatic thrombosis that can contribute to plaque growth.18 Furthermore, thrombectomy specimens from primary percutaneous interventions in ST-elevation myocardial infarction (STEMI) patients show partially organised thrombus in about 50% of the cases suggesting silent lesion progression with thrombus formation prior to the symptomatic acute event.19
Symptomatic thrombosis is responsible for nearly all STEMI incidences and probably about half of non-ST elevation infarcts. The percentage of non-STEMI (NSTEMI) depends somewhat on the definition of the condition and whether you include an elevation of cardiac enzymes immediately post coronary intervention, which is not usually caused by thrombosis. In general, type 1 myocardial infarctions by the universal definition of myocardial infarction are caused by an acute obstructive process in an epicardial coronary artery and this process is usually thrombus formation.20 Thrombus formation in myocardial infarction or sudden cardiac death has been shown to be secondary to three pathologic processes. The most common is plaque rupture followed by plaque erosion (Figure 1). The least common is a calcified nodule, which occurs in <10% of cases.21
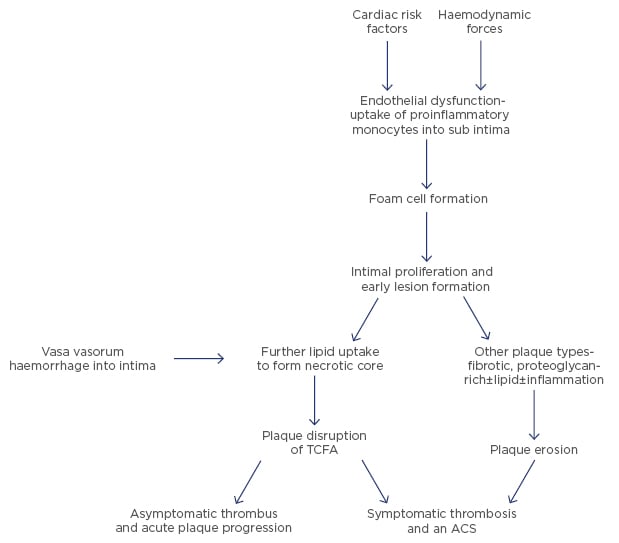
Figure 1: From atherosclerosis to atherothrombosis.
ACS: acute coronary syndrome; TCFA: thin-cap fibroatheroma.
Plaque rupture of a thin cap atheroma is the usual lesion responsible for most thrombotic events, particularly in STEMI. The numbers vary from about 60–75% depending on how the diagnosis was made.22 While pathologic studies originally identified thrombi in those who died suddenly or after myocardial infarction, more recently, intravascular imaging, particularly with optical coherence tomography imaging during primary percutaneous intervention for STEMI or NSTEMI, can differentiate plaque rupture from other mechanisms, including plaque erosion.23
Inflammation is an important mechanism in plaque rupture for a thin cap atheroma with an abundant inflammatory substrate. Thinning and disruption of the cap leading to thrombus formation is precipitated by macrophages and T lymphocytes that are abundant in these lesions. Macrophages also furnish the bulk of the enzymes that catabolise collagen, a key constituent of the fibrous cap of the plaque. Overexpression of interstitial collagenases (MMP-1, MMP-8, MMP-13) in human atheromata are colocalised with macrophages bearing these proteinases.24 Ultimately, thinning of the cap leads to rupture of the cap. Flowing blood is exposed to the under surface of the plaque, which is highly thrombogenic. Platelet deposition is followed by tissue factor activation and the thrombus, if it occludes the lumen (which occurs about 85% of the time in STEMI), will have a platelet-rich head and a red cell and fibrin tail.
Plaque erosion is the second leading pathophysiology for thrombosis in acute coronary syndromes. Pathologic studies suggest that an average of 25–44% of fatal events caused by acute coronary thrombosis are related to erosion.25 Intravascular imaging studies with optical coherence tomography at the time of coronary intervention in myocardial infarction patients have shown erosion as the main mechanism of STEMI in about 25–30%.23 In a limited number of patients interviewed, erosion was more frequent in NSTEMI Type I than in STEMI. However, all these intravascular observations have been performed in small studies after the removal of the thrombus by thrombectomy and have been reported by very few groups. Thus, how well these data can be generalised to acute myocardial infarction is presently unclear.
The role of inflammation in plaque erosions is controversial. Pathologic studies of plaque erosion differ on the role of inflammatory cells adjacent to the thrombus.21,26 While this continues to be debated, inflammation is undoubtedly an important determinant of hypercoagulability, which contributes to thrombus formation in both plaque erosions and plaque disruptions. It is possible to speculate that hypercoagulability of blood may represent a mechanism responsible for inducing or extending the amount of thrombus and thereby converting an asymptomatic thrombus into a symptomatic event.
INFLAMMATION AND HYPERCOAGULABILITY
Inflammation and hypercoagulability promote atherothrombosis. In fact, the complex interplay between inflammation, platelet function, and hypercoagulability is largely responsible for the progression from stable to unstable plaque and an acute coronary event. The effects of inflammation on thrombosis involve both enhanced platelet reactivity, as well as activation of the coagulation cascade from tissue factor expressed by macrophages and other cells. In addition to CRP, other molecules, such as the various IL, TNF, and adhesion molecules, have been associated with increased ischaemic events. For example, IL-6 not only increases CRP, but it also increases platelet reactivity and fibrinogen levels.27 The inflammatory marker, hsCRP, as already discussed, has been shown to predict future thrombotic complications, such as acute myocardial infarction or stroke. Based on all the data, hsCRP appears to be a marker for, and possibly an active participant in, promoting atherothrombotic complications28 (Figure 2).
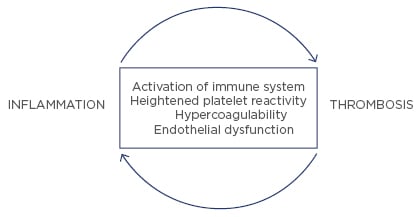
Figure 2: Interrelationship between inflammation and thrombosis.
The relationship between inflammation and thrombosis is bidirectional. Inflammation promotes thrombosis and, vice versa, thrombosis promotes inflammation. Platelet granules contain several proinflammatory chemokines and cytokines.29 Thrombin, a key protein in the coagulation cascade, can also increase the expression of proinflammatory cytokines from vascular endothelial and smooth muscle cells.30 Furthermore, platelets, in experimental studies, exert a host of proatherogenic activities and create a link between haemostasis, innate immunity, atherosclerosis, and inflammation.31
All the standard CAD risk factors are proinflammatory and prothrombotic; they promote endothelial dysfunction and thus interfere with the protective (anti-inflammatory and antithrombotic) effects of nitric oxide. Perhaps the most prothrombotic of these risk factors is exposure to cigarette smoke. The authors and other research groups have published extensively on this subject and a few salient features will be discussed.32,33 Both active and passive cigarette smoke exposure increase the risk of coronary thrombosis and myocardial infarction. According to the National Cardiovascular Data Registry (NCDR), which tracks coronary interventions in all hospitals in the USA, about 45% of STEMI patients undergoing primary intervention occurred in active or recent cigarette smokers.34
Cigarette smoke exposure alters the haemostatic process via multiple mechanisms, which include alteration of the function of endothelial cells, platelets, fibrinogen, and coagulation factors. This creates an imbalance of antithrombotic/prothrombotic factors and profibrinolytic/antifibrinolytic factors that support the initiation and propagation of thrombosis. A growing body of evidence supports free radical-mediated oxidative stress and loss of the protective effect of nitric oxide playing a central role in cigarette smoke exposure-mediated thrombotic diseases.32,35
Smoke exposure increases inflammatory mediators such as TNF-α; CD40 ligand; and the expression of adhesion molecules on endothelial cells, platelets, and monocytes. Inflammation increases tissue factor expression on monocytes and the activated endothelium, as well as activation of circulating tissue factor-bearing microparticles. In humans, chronic cigarette smoke exposure increased levels of multiple inflammatory markers, including peripheral leukocytosis, hsCRP, homocysteine, IL-6, and TNF-α. Exposure of human umbilical vein endothelial cells to cigarette smoke condensate resulted in increased nuclear factor κB DNA binding activity, increased surface expression of intercellular adhesion molecule-1, vascular cell adhesion molecule-1, and platelet endothelial cell adhesion molecule-1. All the above factors are prothrombotic.35
CLINICAL CORRELATIONS
Antithrombotic Agents
Given the importance of thrombosis in myocardial infarction, it is not surprising that antithrombotic agents should have a role both in the prevention and treatment of acute coronary complications. However, there is no clinical evidence that the haemostatic system can attenuate or regress plaque formation. Nevertheless, the importance of antithrombotic agents in coronary disease management was first established in the 1980s when ISIS-236 showed a significant reduction in 30 day mortality with aspirin use alone in patients presenting with acute transmural infarction, and the VA Cooperative study showed that 324 mg of aspirin reduced death and nonfatal myocardial infarction by >50% at 12 weeks in patients admitted with unstable angina.37 Aspirin in low doses irreversibly inhibits COX-1 in the platelet, leading to a significant inhibition of platelet-dependent thromboxane formation, which is responsible for its antithrombotic effect. Aspirin may also inhibit inflammatory cytokines independently of its effect on thromboxane. Its clinical significance is unknown.38
Since the 1980s, antiplatelet drugs have been a mainstay of therapy for medically managed and interventional therapy. Most of the recommendations are in secondary prevention, with limited recommendations for primary prevention; even then, primary prevention is recommended only in higher risk patients. In antiplatelet therapy, the data indicate that the more pathways inhibited in the platelet, the greater the antithrombotic (anti-ischaemic) effect but, at the same time, the greater the bleeding risk.39 Thus, dual antiplatelet therapy is the present class 1 recommendation for stent placement. Use of the more potent ADP receptor blockers, prasugrel or ticagrelor, are more anti-ischaemic with significant reductions in adverse events, such as death, MI, or stroke, compared with the weaker clopidogrel (all with low dose aspirin). However, they are associated with a higher bleeding risk. Likewise, the COMPASS trial40 assessed the role of the low dose anticoagulant, rivaroxaban, plus low dose aspirin in patients with stable cardiovascular disease without a recent stent. While ischaemic events, such as recurrent myocardial infarctions, were significantly reduced on follow up, it is not surprising that bleeding events were also increased.40
Anti-Inflammatory Agents
Up until recently, there was no positive data on specific anti-inflammatory drugs in the management of coronary disease patients. On the other hand, the use of some COX-2 inhibitors, for indications other than heart disease, was associated with increased adverse coronary events.41 Nevertheless, statins possess anti-inflammatory properties, as indicated by a lowering hsCRP, which have been considered to add to their protective effect regarding the reduction of cardiovascular events. As already mentioned, in JUPITER, apparently healthy patients with mild hyperlipidaemia and elevated hsCRP had a significant reduction in major cardiovascular adverse events with rosuvastatin therapy.15
A proof of targeting specifically inflammation to improve outcomes in atherosclerosis was finally accomplished in the CANTOS trial, as previously discussed. Another trial has been in progress targeting the same IL-1β. CIRT is a double blind, randomised trial of low dose methotrexate versus placebo in diabetic or metabolic syndrome patients with a prior history of myocardial infarction who will be followed up for 3–5 years.42 Recently, the Data and Safety Monitoring Board (DSMB) recommended that enrolment in this trial should stop, but not for any substantive safety reasons. The National Heart, Lung, and Blood Institute (NHLBI) halted the trial with about 70% of the 7,000 patients randomised and the results will hopefully be available by the end of 2018. Whether the trial was stopped for futility or because there was a significant benefit with methotrexate is presently unknown.
CONCLUSIONS
Inflammation and thrombosis are important interrelated processes in the development of coronary atherosclerosis and the genesis of atherothrombosis. Thrombosis, while the most common cause of acute myocardial infarction, is also a mechanism for the asymptomatic, non-linear progression of atherosclerosis. Inflammation is paramount in both early and late stages of atherosclerosis. Plaque rupture, the major cause of STEMI and sudden coronary death related to coronary thrombosis, is precipitated by inflammatory-mediated degradation of the thinned fibrous cap overlying lipid-rich plaques.
While antithrombotic agents are an important component of care in the treatment of acute coronary syndromes as well as preventive therapy in high-risk primary prevention and in secondary prevention, the authors have explored the potential role of specific anti-inflammatory agents in the clinical care of these patients. If such therapies (other than statins) are to be considered in the future, anti-inflammatory drugs must significantly reduce events while not adversely overwhelming a patient’s natural immunity to an extent that erases any potential benefit. However, the authors believe that to reduce substantially the morbidity and mortality associated with atherosclerosis, it will be best to interrupt plaque development early in the natural history of atherosclerosis, years prior to any adverse event. This can be accomplished with standard medical therapies as well as lifestyle interventions.43






