Abstract
The introduction of lipid lowering medications was initially thought to provide a solution to the growing burden of coronary heart disease. However, 30 years later, the rates of acute coronary syndrome remain unacceptably high. This realisation forced cardiovascular scientists to look beyond lipids and led to the now widely accepted understanding of atherosclerosis pathobiology: immune-facilitated lipid retention with focal and generalised chronic inflammation. A fundamental component of this inflammatory process is chemokines: a class of cytokines characterised by their ability to facilitate cell recruitment, although it is now known that their function extends beyond chemotaxis. Mounting evidence suggests that chemokines are essential for the destabilisation and subsequent rupture of atherosclerotic plaque. Therefore, chemokine pathways provide a novel therapeutic target for plaque stabilisation. This review addresses the role of chemokines in regulating plaque vulnerability and discusses therapeutic approaches targeted at manipulating chemokine pathways.
INTRODUCTION
In the late 20th century, lipid lowering medications appeared to be the answer for coronary heart disease. However, despite reductions in mortality, the incidence of acute coronary syndrome (ACS) still remains unacceptably high.1The last decade has seen a significant shift in the understanding of atherosclerosis pathobiology; the disease is now widely accepted as a product of immune-facilitated lipid retention with focal and generalised chronic inflammation.2 Furthermore, it is known that certain features of atherosclerotic plaque determine its vulnerability, and therefore its susceptibility to rupture. With this enhanced understanding comes a new generation of therapies targeted at the inflammatory response. Nevertheless, manipulation of the immune system is not without its challenges and the optimal target for anti-inflammatory drugs remains elusive. This review aims to address the role of chemokines in regulating plaque vulnerability and discuss therapeutic approaches targeted at manipulating chemokine pathways.
CHEMOKINES
Chemokines are a class of cytokines characterised by their ability to exert chemotactic effects, although their functionality does not stop there. Chemokines are divided into four subclasses by virtue of their N-terminal cysteine residue arrangement: C, CC, CXC, and CX3C.3 Chemokines act via a family of class A G-protein-coupled receptors, collectively termed chemokine receptors. Commonly, a single chemokine ligand is shared by a number of receptors, for example, CCL5 interacts with CCR1, CCR3, and CCR5. Multiple cell types secrete chemokines and express their cell surface receptors. In the setting of atherosclerosis, white blood cells (WBC), platelets, endothelial cells (EC), and smooth muscle cells (SMC) contribute the vast majority of chemokine ligands and receptors.3
VULNERABLE PLAQUE
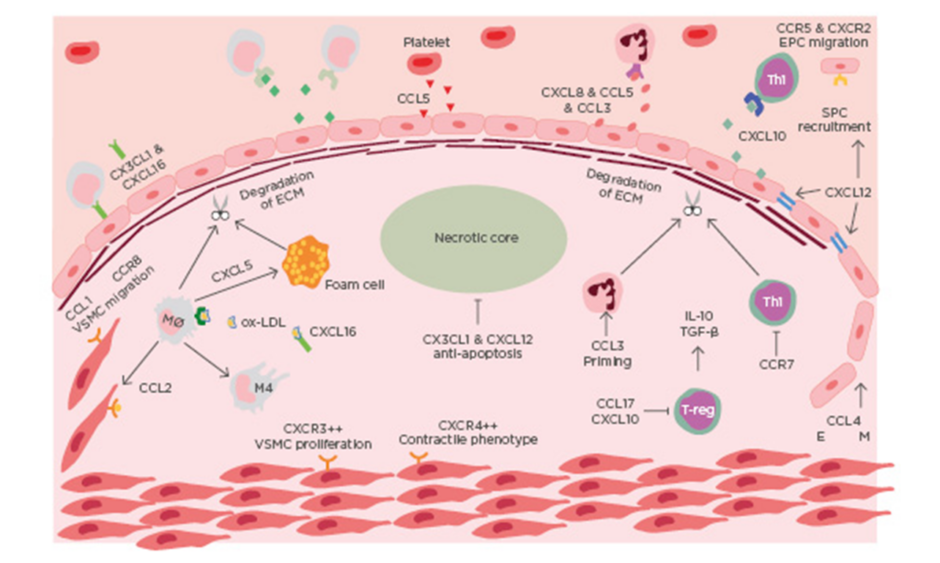
ACS is a clinical manifestation incorporating both unstable angina and myocardial infarction, with and without ST-segment elevation. Most commonly ACS is preceded by rupture of a vulnerable coronary plaque leading to thrombosis and subsequent occlusion of blood flow.4 There is increasing evidence that suggests plaque stability is regulated by chemokines (Table 1). Chemokines regulate plaque stability by modulating immune cell infiltration, systemic inflammation, fibrous cap thickness, necrotic core size, collagen content, and SMC accumulation (Figure 1).
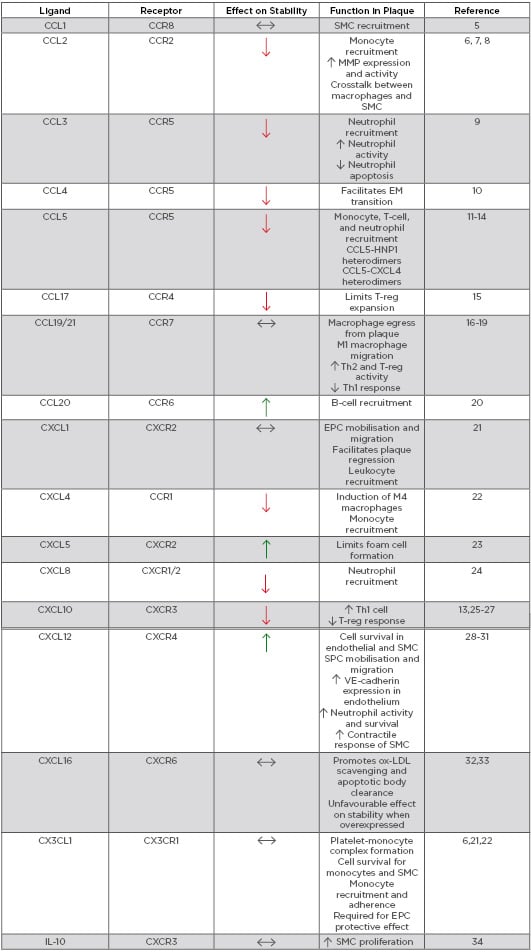
Table 1: Role of chemokines in modulating plaque stability.
EM: endothelial-mesenchymal; EPC: endothelial progenitor cell; HNP1: human neutrophil peptide 1; IL-10: inducible protein-10; MMP: matrix metalloproteinase; ox-LDL: oxidised low-density lipoprotein; SMC: smooth muscle cell; SPC: smooth muscle progenitor cell; Th: T-helper cell; T-reg: regulatory T-cell; VE-cadherin: vascular endothelial cadherin.
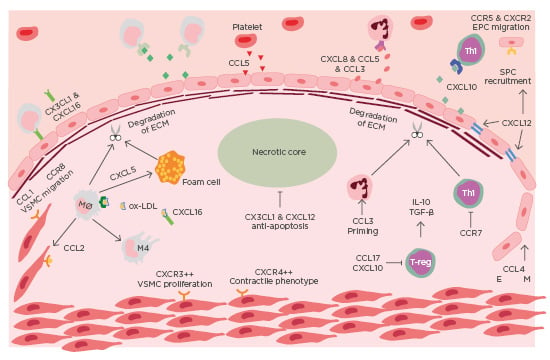
Figure 1: Chemokines in the destabilisation of atherosclerotic plaque.
Immune cells are recruited to atherosclerotic lesions by chemokines, primarily produced by the dysfunctional endothelium. Platelets also contribute to the chemotactic gradient by secreting CCL5. The secretion of MMP from intraplaque immune cells leads to the degradation of collagen. The necrotic develops as a result of impaired efferocytosis. Endothelial-mesenchymal transition occurs in advanced lesions, mediated by CCL4. EM: endothelial mesenchymal transition; EPC: endothelial progenitor cell; mono: monocyte; Mø: macrophage; Neu: neutrophil; ox-LDL: oxidised-low-density lipoprotein; TGF-β: transforming growth factor β; Th1: T-helper cell 1; VSMC: vascular smooth muscle cell.
IMMUNE CELL INFILTRATION
Immune cell infiltration is a hallmark pathological feature of atherosclerosis. Although monocytes/macrophages are the primary immune cell linked to atherosclerosis, plaque stability is the product of a complex interplay between several WBC including monocytes, macrophages, neutrophils, and lymphocytes.2 Recruitment of WBC to atherosclerotic lesions is driven by a chemotactic gradient, produced largely by a dysfunctional endothelium. Once recruited, many cells become trapped in the lesion and secrete a plethora of proinflammatory cytokines. Prolonged inflammation ultimately degrades plaque structure and renders the lesion susceptible to rupture.2
Monocytes/Macrophages
Circulating monocytes demonstrate heterogeneity by differential expression of chemokine receptors. Plaque progression relies heavily upon the continual recruitment of monocytes and, unsurprisingly, plaque vulnerability is closely related to macrophage content. Classical monocytes, which account for up to 90% of circulating monocytes, typically employ CCR2 and CX3CR1 to migrate into plaque.35 On the other hand, non-classical monocytes primarily use CCR5.35 Deficiency in any one of the aforementioned chemokine receptors or their respective ligands results in a substantial decrease in monocyte recruitment to plaque. On the other hand, combined inhibition of CCL2, CX3CR1, and CCR5 completely inhibits intraplaque macrophage accumulation.36 These three chemokine axes work together to direct monocyte recruitment to sites of atherosclerotic lesions; hence, CCL2, CCL5, and CX3CL1 are all highly expressed in vulnerable plaque.37
Although monocyte recruitment appears largely dependent upon activation of CCR2, CCR5, and CX3CR1, inhibition of other chemokine pathways also suppresses macrophage accumulation within plaque. This suggests that activation of the primary chemotactic pathways requires functional secondary interactions. For example, activated platelets deposit CCL5 on the surface of EC which interacts with HNP1, secreted from neutrophils, to promote monocyte adhesion. Blocking this HNP1–CCL5 heterodimer stunts myocyte recruitment.14 A similar heterophilic interaction occurs between CCL5 and CXCL4, which, when inhibited, suppresses monocyte infiltration.12
The chemokines CXCL16 and CX3CL1 are unique multifunctional proteins that can be both expressed on the cell surface or cleaved to act as a soluble ligand. Membrane-bound CX3CL1 facilitates monocyte adhesion to an inflamed endothelium and the formation of platelet-monocyte complexes that are subsequently recruited to plaque.38 The role of CXCL16 is more elusive. Deficiency of CXCL16 in mice increases plaque size, which is thought to be the result of reduced oxidised-low-density lipoprotein scavenging and apoptotic body clearance.32 Conversely, CXCL16 overexpression promotes plaque destabilisation.33
In humans, high levels of soluble CXCL16 is prognostically unfavourable in ACS patients.32 This discrepancy may be the product of differing effects of membrane-bound versus soluble CXCL16.
CXCL5 does not directly recruit monocytes to the dysfunctional endothelium; nevertheless, it is involved in macrophage trapping within plaque. Inhibition of CXCL5 increases monocyte-derived foam cell accumulation in plaque. Analogously, exogenous administration of CXCL5 limits foam cell formation. This effect is as a result of CXCL5/CXCR2 signalling, which improves cholesterol efflux from macrophages by upregulating ABCA1 expression.23 In humans, there is a strong negative association of circulating CXCL5 levels with coronary artery disease severity.39
Lymphocytes
T cells are a heterogenous population with many subtypes, several of which are involved in atherosclerosis. Th1 cells are the most abundant T cell in human atherosclerotic plaque. IFN-gamma is the prototypic cytokine secreted from these cells and is known to promote plaque vulnerability by impairing collagen formation and maturation.40 Th1 cells express high levels of CXCR3, which is required for their migration into atherosclerotic plaque.25 Antagonism of CXCR3 or its ligand CXCL10 alters the T-cell response to an atherogenic stimulus by reducing Th1 cell infiltration and increasing regulatory T-cell (T-reg) accumulation.26 Inhibition of the CXCL10/CXCR3 axis produces a more stable plaque phenotype and in humans, high CXCL10 expression is predictive of vulnerable plaque.27 Moreover, dual inhibition of CCR5 and CXCR3 suppresses the T-cell response in atherosclerosis, by reducing T-cell accumulation and INF-gamma expression by 95% and 98%, respectively.13
CCR7 is another chemokine receptor involved in regulating the T-cell response. CCR7 knockout exacerbates atherosclerotic disease progression.
CCR7-deficient mice display enhanced T-cell accumulation in plaque and polarisation of the T-cell response, whereby serum levels of IL-4 and TGF-β decrease and IL-12p40 and IL-17F increase. This implies that CCR7 signalling is responsible for maintaining the Th2 and T-reg response, while suppressing Th1 activity.16 In parallel, CCR7 expression in peripheral blood mononuclear cells is significantly reduced in patients with unstable coronary disease compared to those with a stable disease phenotype. Treatment of these patients with aggressive statin therapy has been shown to enhance CCR7 expression.17
T-regs are important negative regulators of the T-cell response. Naturally, T-regs account for 1–5% of the total lesional T cells. CCL17, a dendritic cell-derived chemokine, is known to limit T-reg expansion. CCL17 deficiency enhances T-reg expansion, increases T-reg intraplaque accumulation, and attenuates atheroma progression. Additionally, atherosclerotic lesions in CCL17-deficient mice, display a more stable phenotype with increased SMC content. These findings are supported by the elevated expression of CCL17 observed in human atherosclerotic tissue.15 Similar to T-regs, B-cells appear to be atheroprotective. Global B-cell deletion exacerbates the development and progression of atherosclerosis in mice.20 B-cells are recruited to plaque via CCL20/CCR6 and adoptive transfer of CCR6+/+ B-cells to ApoE-/- mice induces plaque regression.20
Neutrophils
More recently, neutrophils have been identified as key players in plaque instability. Activation of neutrophils results in the release of preformed inflammatory proteins and ultimately the generation of neutrophil extracellular traps, which in turn drive plaque rupture.4 Neutrophils naturally express CCR5, which can be increased by the presence of a high-fat diet.9 It has been demonstrated that platelet-derived CCL5 acts via CCR5 to induce neutrophil migration into plaque.11 CCL5 is also known to interact with CXCL4 and specific blocking of this interaction reduces neutrophil recruitment and neutrophil extracellular traps formation.12 Furthermore, CXCL8, produced largely by monocytes and macrophages, promotes lesional neutrophil accumulation via CXCR1 and CXCR2. In human subjects with stable coronary artery disease, baseline serum CXCL8 is a robust predictor of future cardiovascular events.24
The chemokine receptors CXCR2 and CXCR4 function in an opposing fashion to regulate neutrophil trafficking. CXCR2 is required for neutrophil recruitment from the bone marrow. In contrast, activation of CXCR4 promotes neutrophil retention in the bone marrow.41 Mice treated with a CXCR4 antagonist display a 4-fold increase in relative neutrophil accumulation within plaque42 and a higher incidence of intraplaque haemorrhage.28 Neutrophils lacking functional CXCR4 have augmented myeloperoxidase activity and enhanced cell survival, illustrating that CXCR4 deficiency not only increases neutrophil homing to plaque, but also amplifies their activity.28 CCL3 plays a similar role in neutrophil homeostasis. Haematopoietic deficiency of CCL3 reduces both circulating and plaque neutrophil content, which is most probably attributable to higher rates of apoptosis observed in CCL3-deficient neutrophils. Additionally, the absence of CCL3 impairs neutrophil responsiveness to other chemotactic stimuli, such as CXCL1.9
Chemokines and the Necrotic Core
Generation of the necrotic core results from impaired efferocytosis: a combination of profound intraplaque apoptosis and reduced phagocytic capacity of macrophages. Chemokines have direct and indirect effects on the necrotic core. Indirectly, chemokines mediate immune cell infiltration and thus, the extent of intraplaque apoptosis. Directly, chemokines are involved in cell survival signalling and macrophage polarisation. Treatment of mice with CX3CL1-fc, a long acting CX3CR1 agonist, reduces necrotic core burden, at least partly due to the antiapoptotic effects of CX3CL1 signalling in macrophages and SMC.43 CXCL12 is known to have a similar effect, whereby CXCL12/CXCR4 signalling counteracts apoptosis in endothelial and SMC.29
Macrophage populations differ in their response to various chemotactic stimuli and their phagocytic ability. M1 macrophages migrate in response to CCL19/CCL21 signalling via CCR7. Although M2 macrophages express similar amounts of CCR7 they do not respond to CCL19/CCL21 stimulation.18 CCL19/CCL21 signalling has also been implicated in macrophage egress from lesions. Inhibition of both ligands suppresses plaque regression and it has been proposed that the macrophage egression properties of statins may be CCR7-dependent.19 The platelet-derived chemokine, CXCL4, has recently been shown to induce a novel macrophage phenotype, termed M4. M4 macrophages display a pro-inflammatory phenotype, characterised by high IL-6, TNFα, and matrix metalloproteinase-7 (MMP-7) expression, in combination with reduced phagocytic capacity. High intraplaque accumulation of M4 macrophages is associated with features of vulnerability.22 This indicates that platelet activation may promote progression of the necrotic core via the secretion of CXCL4.
CHEMOKINES AND THE FIRBROUS CAP
The fibrous cap is typically a collagen-rich structure interspersed with SMC that acts as a barricade between the pro-thrombotic core of a lesion and the circulating coagulation factors. Neointimal recruitment of SMC is facilitated by CCL1 and CCR8.5 Continual SMC recruitment in combination with the deposition, cross-linking, and maturation of collagen leads to the formation of the fibrous cap. The thickness, and therefore stability, of the cap is largely dependent on two factors: MMP expression and SMC content. Rupture-prone regions of human atherosclerotic plaque show increased levels of MMP 1, 3, 8, 9, 11, 14, and 16, in combination with reduced SMC accumulation.44
There is extensive evidence describing a regulatory effect of chemokines on MMP activity. Stimulation of monocytes with either CCL2, CCL3, CCL4, or CCL5 increases the expression of MMP-9 and MMP-14.6,7 It is thought that these effects are primarily mediated via CCR1 and CCR2, as mice deficient in these receptors have diminished MMP production.45,46 Additionally, CCL2 is known to mediate cross-talk between macrophages and SMC, particularly in a high glucose environment. In this setting, macrophage-derived CCL2 increases MMP-1 and MMP-9 secretion from SMC.8
Chemokines modulate SMC accumulation and functionality in plaque. Mice deficient in CCR5 show increased SMC and IL-10 intraplaque expression, suggesting that CCR5 may be a negative regulator of SMC migration into plaque.47 Alternatively, activation of CXCR3 enhances SMC proliferation.34 Similarly to many of the cells involved in atherosclerosis, SMC demonstrate plasticity, which can be influenced by CXCR4. SMC-specific CXCR4-deficiency causes a phenotypic switch from contractile to synthetic: a switch associated with foam-cell formation.30 Smooth muscle progenitor cells, when recruited to atherosclerotic lesions, promote stability. CXCL12 treatment mobilises smooth muscle progenitor cells from the bone marrow and recruits them to atherosclerotic plaque. Mice treated with CXCL12 display increased fibrous cap thickness.31 In humans, plasma CXCL12 expression is reduced in patients with coronary heart disease, particularly in those with an unstable phenotype.48 This reduction in CXCL12 may produce the upregulation of the CXCL12 receptor, CXCR4, seen in patients with unstable lesions.49
Endothelial Integrity
Plaque erosion, characterised by endothelial denudation at the plaque-blood interface, is another mechanism that promotes instability. Endothelial-mesenchymal transition, a process whereby EC polarise toward a myofibroblast-like cell and accumulate within plaque, is known to alter plaque stability. The extent of endothelial-mesenchymal transition is greatly increased in unstable and ruptured plaques.10 Yang et al.50 demonstrated that foam cell-derived CCL4 acts via EC-expressed CCR5 to promote endothelial-mesenchymal transition in atherosclerotic plaque: a process that could be inhibited by maraviroc, a commonly available CCR5 antagonist. Activation of CXCR4 by its ligand CXCL12 provides an antiatherogenic effect by enhancing endothelial barrier function. More specifically, CXCL12/CXCR4 signalling increases vascular endothelial cadherin expression, therefore stabilising endothelial junctions.30
Endothelial progenitor cells (EPC) are important for maintaining a functional endothelium. Patients with atherosclerosis have reduced EPC numbers and enhancing EPC activity has been shown to promote plaque regression.51 EPC recruitment to plaque is driven by the activation of CCR5 and CXCR2. CCR5 overexpression in mice, dramatically increases lesional EPC recruitment, EC coverage of coronary plaque and systemic nitric oxide levels; indicating improved EC function.21 Similarly, CXCR2 antagonism blunts EPC migration and EPC induced plaque regression. In the same study, it was found that CX3CR1 expression was required for the atheroprotective effects of EPC, despite the fact that CX3CR1-deficiency did not alter EPC migration.52
THERAPEUTIC CHEMOKINE MODULATION TO ENHANCE PLAQUE STABILITY
Plaque stability is largely influenced by inflammation. As such, immunotherapies provide a novel method for promoting stability of atherosclerotic lesions. Results from the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS)53 confirmed that specifically targeting components of the inflammatory response can be effective in the secondary prevention of cardiovascular disease. However, immunotherapies have a common pitfall: potential oversuppression of the immune system, rendering the host susceptible to infection.53 This adverse effect is the primary barrier stalling the large-scale implementation of immunotherapies in patients with atherosclerosis. Proper immune function is a delicate balance between infection (underactivation) and inflammation (overactivation). Therefore, finding the ideal therapeutic target that does not offset this equilibrium remains key to developing effective immunotherapies.
As described above, chemokines are an essential component of the inflammatory response, ultimately responsible for the destabilisation of plaque. Consequently, chemokine-targeted therapies have gained popularity in recent years. Numerous preclinical studies have demonstrated that antichemokine therapies are effective at reducing plaque burden and instability. Despite this, there is a lack of translational data confirming these findings in patients with atherosclerotic disease.
CCL2/CCR2
The CCL2/CCR2 axis is the most commonly targeted chemokine pathway in atherosclerosis. Generally speaking, inhibition of this pathway stunts progression and promotes a morphologically stable plaque. Local gene silencing of CCL2 reduces plaque disruption rates by approximately 80%, in mice. This improved stability was facilitated by increased fibrous cap thickness and SMC and collagen content, in combination with a large reduction in lesional macrophage accumulation.54 Antagonism of CCR2 produces similar results. Bot et al.55 reported that administration of 15a, an orthosteric small molecule CCR2 antagonist, greatly reduced intraplaque macrophage accumulation and necrotic core size. 15a also reduced circulating classical monocytes number, suggesting that CCR2 is required for classical monocyte recruitment from the bone marrow.55 On the contrary, a reduction in circulating monocyte numbers may imply an impaired immune response. Another CCR2 antagonist, 7ND, has been trialled in mice and rabbits. Treatment of ApoE-/- mice with 7ND increased the plaque stability score, as measured by collagen, macrophage, and lipid composition of the lesion.56 In rabbits, administration of 7ND prevented plaque disruption, which was associated with an increase in fibrous cap thickness.57 Evidently, inhibition of the CCL2/CCR2 axis is effective at suppressing the atherogenic immune response. However, recent evidence suggests that the efficacy of such treatment is related to circadian rhythm and thus, dosing is time dependent. Winter et al.58 demonstrated that myeloid cell recruitment from bone marrow is not constant; it occurs in peaks throughout the day. As such, the authors further reported that CCR2 neutralisation is most effective when administered just prior to the peak of myeloid cell recruitment.58
To the authors’ knowledge, there has only been one CCR2 antagonist trialled in humans. Gilbert et al.59 employed MLN1202, a CCR2-specific monoclonal antibody, to assess the effect of CCR2 blockade on C-reactive protein levels in individuals at increased risk of atherosclerosis. This study demonstrated that short-term CCR2 blockade was not only safe but efficacious in suppressing atherosclerosis-associated inflammation. Larger Phase III trials are required to confirm these findings and evaluate long-term safety and efficacy of CCR2 antagonism in humans with atherosclerotic disease.
CX3CR1
Many of the methods employed to disrupt chemokine pathways are not easily translatable to human clinical trials. To address this, Zhou et al.60 developed an anti-CX3CR1 vaccination, which was trialled in ApoE-/- mice. Vaccinated mice showed a 35% reduction in plaque size and a 5-fold reduction in macrophage infiltration. Despite this, the M1/M2 lesional macrophage ratio or SMC content was unaltered by vaccination.60 In direct contrast, Riopel et al.43 reported that enhanced CX3CR1 activation is atheroprotective, whereby, administration of CX3CL1-fc to mice produced a more stable plaque. CX3CL1-fc treated mice had reduced M1 and T-cell accumulation and a smaller necrotic core. Moreover, these mice had enhanced intraplaque SMC and collagen content.43
CCL5/CCR5
The CCL5/CCR5 axis has also been an attractive target for modulating plaque. Met-RANTES, a CCR1 and CCR5 antagonist, displays a significant atheroprotective effect. LDLr-/- mice treated with Met-RANTES show reduced macrophage and lymphocyte infiltration and reduced MMP-9 expression. Consequently, these lesions had a thicker fibrous cap and increased intraplaque collagen.61 Similar effects were reported with the use of [44AANA47]-RANTES, a CCL5 antagonist. Administration of [44AANA47]-RANTES promoted regression of established lesions and produced a more stable plaque.62 The CCR5 antagonist maraviroc, also appears effective at stunting plaque progression. In a mouse model of late-stage atherosclerosis, daily administration of maraviroc attenuated plaque progression and intraplaque macrophage accumulation.63
Other Chemokine Targets
Several other chemokine ligands and receptors have been targeted in atherosclerosis. Administration of a CXCL10-neutralising antibody has been shown to prevent vulnerable plaque development by increasing lesional SMC and collagen content, while significantly reducing the relative necrotic core size.27 Evasin-3, a CXCL1 and CXCL2 antagonist, promotes plaque stabilisation by reducing intraplaque neutrophil infiltration and MMP-9 expression.64 Antagonism of CXCR3 or CCL17 may also promote plaque stabilisation by increasing T-reg expansion. Inhibition of either receptor causes an increase in T-reg expansion and a reduction in plaque progression.15,26
A group of scavenger receptors involved in the regulation of chemokine activity have recently been described in the literature, termed atypical chemokine receptors (ACKR). ACKR differ from classical chemokine receptors as they do not couple G proteins; thus, they act to scavenge, internalise, and transport chemokines without initiating a signalling cascade.65 There are currently four known ACKR, two of which are clearly involved in the progression of atherosclerosis. ACKR1, expressed primarily on erythrocytes and venular EC, binds >20 different inflammatory chemokines. ACKR1-deficiency in mice stunts atherogenesis and skews macrophage polarisation toward a less inflammatory phenotype.66 ACKR1-deficiency has also been shown to suppress macrophage migration by inhibiting CCL2 expression on the surface of erythrocytes.67 In contrast, ACKR3 appears to be atheroprotective. Genetic ablation of ACKR3 increases systemic cholesterol levels and promotes progression of atherosclerotic lesions.68 Moreover, ACKR3 is involved in mediating EPC survival and homing as well as vascular SMC migration.65 Pharmacological manipulation of ACKR expression may provide a novel mechanism to regulate chemokine activity and minimise global immune suppression. Therefore, future studies investigating the possibility of pharmacological ACKR modulation would be of great benefit to cardiovascular research.
CURRENT AND FUTURE DIRECTIONS
Despite abundant preclinical data, there is limited translational research investigating the effect of antichemokine medications in patients with atherosclerotic disease. This may be due to the potential increased risk of infection and/or the enormous cost associated with drug development. However, results from oncological studies suggest otherwise. Many antichemokine therapies have been trialled in cancer patients and demonstrate a sound safety and tolerability profile.69 Adapting these medications for use in cardiovascular disease may allow cost and time-effective development of antiatherogenic, chemokine-targeted therapies. Future studies should aim to translate lab-based findings into robust clinical trials, potentially drawing from the methodology employed in the CANTOS study.53
CONCLUSION
Chemokines are unequivocally involved in all stages of atherosclerosis. Evidently, the role of chemokines extends well beyond the sole recruitment of immune cells and there are now abundant data implicating chemokines in the destabilisation of atherosclerotic plaque. Manipulation of chemokine pathways represents a novel therapeutic target for the stabilisation of coronary plaque; nevertheless, is not without its challenges. Careful selection of drug targets may help mitigate risk and ultimately produce an effective antichemokine therapy.