Abstract
This literature review provides insights into how glutathione (GSH) plays an important role in controlling HIV-1 and Mycobacterium tuberculosis infections. Since the discovery of HIV in 1981, >40 million affected individuals have died due to AIDS, and currently 40 million people are infected with HIV worldwide, which primarily infects CD4+ T cells. The natural pathogenesis of HIV consists of three stages: 1) the primary HIV infection phase, 2) the asymptomatic chronic phase, and 3) the late HIV symptomatic phase, which leads to an immunocompromised state resulting in increased susceptibility to opportunistic infections. It has been shown that HIV+ individuals have low levels of GSH; increased levels of proinflammatory cytokines, which correlate with increased production of reactive oxygen species and oxidative stress; and increased levels of TGF-β compared to healthy individuals. Consequently, increased reactive oxygen species levels lead to decreased levels of reduced GSH and increased levels of TGF-β, which has been demonstrated to inhibit the rate-limiting enzyme responsible for the de novo synthesis of GSH. In addition, the authors demonstrate that with supplementation of reduced GSH, there is improved intracellular control of an M. tuberculosis infection within macrophages. Therefore, decreased levels of GSH can leave HIV+ individuals prone to such opportunistic infections. The HIV transactivator of transcription (TAT) protein has also been shown to further increase oxidative stress and reduce GSH levels. Liposomal-GSH supplementation has the ability to bypass de novo GSH synthesis and provide protection against HIV and M. tuberculosis infections by increasing levels of GSH, improving redox homeostasis, and dampening the effects of TGF-β.
INTRODUCTION
Since the discovery of HIV in 1981, >40 million affected individuals have died due to AIDS. Currently, approximately 40 million people worldwide are living with HIV1 and two-thirds of these reside in Africa, where disease prognosis is poor due to limited access to high-quality healthcare and drugs.2 HIV infection leads to a deficiency in CD4+ T cells and eventual loss of immunity, causing increased susceptibility to opportunistic pathogens. Mycobacterium tuberculosis infection is the most common opportunistic infection in HIV-infected individuals, affecting approximately one-third of HIV patients and accounting for 26% of AIDS-related deaths.3 HIV-1 primarily infects and destroys the Th1 subset of CD4+ T cells, resulting in compromised production of IL-2, IL-12, and IFN-γ, and increased susceptibility to opportunistic infections. Specifically, IFN-γ activates macrophages, neutrophils, and dendritic cells (DC) to destroy intracellular pathogens by producing reactive oxygen species (ROS), reactive nitrogen species, and antimicrobial peptides. IFN-γ can also activate CD8+ T cells and natural killer (NK) cells to produce perforin and granzymes, inducing programmed cell death of the infected target cells and destruction of the intracellular pathogen. Loss of IFN-γ can therefore dampen the ability of the host immune system to control intracellular infections. Glutathione (GSH) enhancement in a concentration-dependent manner (millimolar concentrations) in DC is directly associated with IL-12 production, which in turn favours a Th1 CD4+ T cell response, resulting in the production of IL-2, IL-12, and IFN-γ.
HIV PATHOGENESIS
HIV first comes in contact with and enters the body through a mucosal surface and infects the macrophages and DC present in the mucosal layer at this site of exposure. Here, the viral cell population increases, forming an important latent reservoir of viral cells.4-6 During HIV infection, NK cells induce maturation of DC and initiate DC migration to the lymph nodes. HIV alters DC intracellular mechanisms to prevent NK cell-mediated apoptosis by upregulating cellular FLICE-like inhibitory protein (c-FLIP) and IL-10, thereby mediating protection; this allows for increased HIV viral replication. Concurrently, DC migrate to the lymph nodes where HIV migrates intracellularly towards the tips of the DC projections and is transferred to CD4+ T cells.5 Viral glycoproteins gp120 and gp41 both play a critical role in HIV pathogenesis; when HIV-1 first comes into contact with the host cell, gp120 binds to the host via CD4 molecules expressed on CD4+ T cells and macrophages. Once gp120 binds to CD4, the chemokine receptor type 4 (CXCR4) is expressed, allowing gp120 to bind to it, which is followed by a fusion of gp41 with CXCR4. The viral RNA is then transferred into the CD4+ T cells (Figure 1).
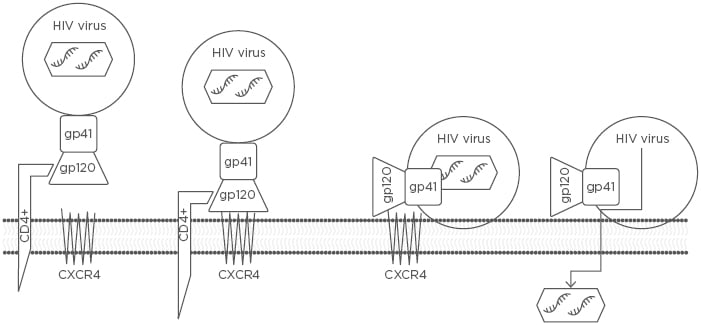
Figure 1: The three steps by which an HIV RNA-containing capsid enters a host CD4+ cell to be replicated.
The three steps include 1) attachment of the HIV envelope gp120 to the CD4+ T cell receptor ligand, 2) gp120 binding to CXCR4, and 3) gp41-mediated fusion of the HIV viral envelope with the host cell membrane via CXCR4, resulting in the transfer of HIV RNA into the CD4+ T cell.
CXCR4: chemokine receptor type 4.
HIV-1 is an RNA retrovirus, meaning that after entering the host cell the viral RNA is transcribed into complementary (c)DNA using reverse transcriptase. Viral cDNA is then incorporated into the host DNA. Preliminary evidence suggests that the binding of CD4 molecules to gp120 on the virus is dependent on the redox state of CD4 and monomeric reduced CD4 is the preferred state. Further studies are required to elucidate the role of GSH in this process.7 A clear understanding of the underlying mechanism could lead to the development of therapeutics that can prevent HIV binding to CD4 molecules.
HIV infection causes a gradual decrease in CD4+ T cell count over time. Once the CD4+ T cell count drops below the threshold value of 200 cells/µL, opportunistic infections such as tuberculosis (TB) are increasingly likely to occur due to immunodeficiency. The natural history of HIV infection progresses in a three-stage process: 1) primary HIV-1 infection or the acute phase, 2) the chronic or latent phase of asymptomatic HIV infection, and 3) the late HIV disease phase.8 In the acute stage, most patients experience symptoms of infectious mononucleosis, characterised by symptomatic fever, rash, and lymphadenopathy. There is also a transient increase in the viral load; however, eventually the HIV viral load is curtailed during this phase due to an HIV-specific CD8+ cytotoxic T cell response. CD8+ T cells produce perforin and granzyme to provide protection against viral infection, resulting in a decline in the viral load.9 During the latent phase, the CD4+ cell count continues to decline while the viral count rises but the patient usually remains asymptomatic. This latent stage can last as long as 10–15 years among HIV patients and, while the patient is clinically asymptomatic, the virus is exceptionally versatile. Consequently, the viral cell count continues to increase inside the lymphoid organs with little or no immune response. During the late HIV disease stage, the CD4+ cell count drops to <200 cells/µL and patients are diagnosed with AIDS. This stage is characterised by the broadening of HIV tropism, resulting in an ever-greater rate of CD4+ T cell loss.8
GLUTATHIONE SYNTHESIS
GSH (γ-glutamylcysteinylglycine) is an important thiol tripeptide antioxidant that maintains cellular redox state and homeostasis in a variety of processes within mammalian cells, particularly those involved in the immune system (Figure 2).10,11 GSH is present in the cytosol of cells in the range of 1–10 mM and 1–3 µm in the plasma.11,12 GSH exists in two forms: the thiol-reduced (rGSH) form and the disulfide-oxidised (GSSG) form, with rGSH accounting for >98% of the total GSH.13 The de novo synthesis of GSH uses glutamine, cysteine, and glycine, and takes place in two steps, both requiring ATP.10 The first step in the biosynthesis of GSH is rate-limiting and is catalysed by glutamate-cysteine ligase (GCL).10 GCL is composed of two subunits: a heavy, catalytic subunit (GCLC) and a light, modifier subunit.10,14 The light, modifier subunit provides a regulatory function, while GCLC is responsible for the catalytic activity, which conjugates sulfur-containing cysteine with glutamate, forming γ-glutamylcysteine.10 GSH synthetase catalyses the second and final step in the de novo synthesis of GSH through the conjugation of glycine with γ-glutamylcysteine.5 GSH exerts a negative feedback inhibition on GCLC.10,14 The regulation of GCL expression and activity is critical for the homeostasis of GSH.10
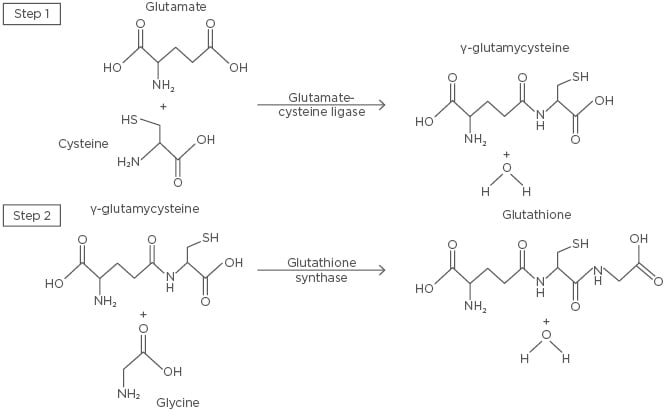
Figure 2: The de novo synthesis of glutathione.
The first and rate-limiting step of glutathione synthesis involves the conjugation of glutamate and cysteine by glutamate-cysteine ligase, forming γ-glutamylcysteine. Glycine and γ-glutamylcysteine are then conjugated by glutathione synthase in the second step, forming glutathione.
GLUTATHIONE AND OXIDATIVE STRESS
ROS contain partially reduced O2 and are produced as biproducts of normal cellular respiration and during enzymatic reactions. ROS have beneficial effects on several physiological processes, including the destruction of pathogens and tissue repair;15 however, when there is a disproportionately high amount of ROS, it induces oxidative tissue damage. ROS are produced in response to a variety of stimuli, such as ultraviolet radiation, cigarette smoke, alcohol, nonsteroidal anti-inflammatory drugs, and infections.15
ROS include radical compounds, such as superoxide, hydroxyl radicals, and lipid hydroperoxides, and reactive nonradical compounds, such as singlet oxygen, hydrogen peroxide (H2O2), hypochlorous acid, chloramines, and ozone.16 These molecules induce oxidative damage through unpaired valence-shell electrons that are highly unstable and reactive and have the ability to disrupt macromolecules, including proteins, lipids, carbohydrates, and nucleic acids.15
In the presence of ROS, rGSH is converted to GSSG by glutathione peroxidase (GPx) (Figure 3).15 GPx links two GSH molecules through a disulfide bridge to form GSSG and, during this process, H2O2 is reduced to water and lipid hydroperoxides to their corresponding alcohols.15 GSSG is unable to perform antioxidant functions but can be converted back to rGSH by glutathione reductase (GSR),15,17,18 which requires the oxidation of NADPH to NADP+.18 GPx and GSR play critical roles in protecting cells from the harmful effects of peroxide decomposition; however, the GSH antioxidant system can be overwhelmed if ROS are produced in excess, leading to increased levels of free radicals that can damage molecules that are essential to cellular homeostasis and metabolism.19
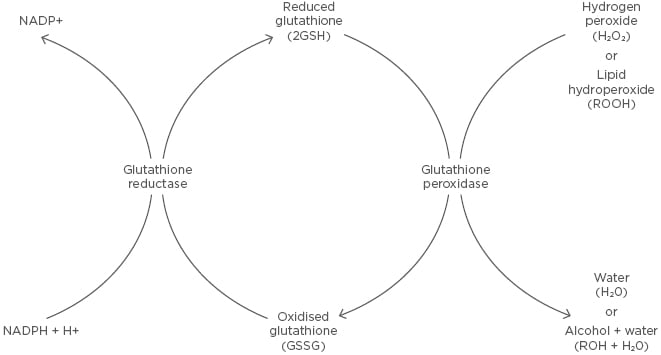
Figure 3: Glutathione redox reaction.
Reduced glutathione is converted to oxidised glutathione through a disulfide bridge by glutathione peroxidase in the presence of hydrogen peroxide or a lipid hydroperoxide, forming water or an alcohol and water, respectively. Oxidised glutathione is converted to reduced glutathione through glutathione reductase, which requires the oxidation of NADPH to NADP+.
DECREASED GLUTATHIONE IN HIV+ INDIVIDUALS
It has been shown that HIV-infected individuals have diminished levels of GSH in their red blood cells, macrophages, T cells, and NK cells.20,21 The authors have demonstrated that within macrophages of HIV+ individuals, GSSG was more heavily favoured and decreased levels of rGSH correlated with a significant increase in the growth of M. tuberculosis.21 In addition, there were also increased levels of proinflammatory cytokines, such as IL-1, TNF-α, and IL-17, which correlated with an increased production of free radicals. There was also a significant increase in the levels of TGF-β in these individuals. TGF-β is involved in regulating a variety of aspects of host defence to injury; however, when it is overexpressed it can contribute to pathogenic manifestations. Increased levels of TGF-β have been demonstrated to decrease expression of GCLC, essential for the rate-limiting step of the de novo synthesis of GSH.21-24
Furthermore, in previous studies it was demonstrated that GSH is essential for controlling the intracellular concentration of M. tuberculosis within macrophages.25 In a later study, it was shown that in vitro treatment of whole blood cultures from HIV+ subjects with N-acetyl cysteine (NAC), a GSH precursor, resulted in improved control of intracellular M. tuberculosis infection, as well as decreased levels of proinflammatory cytokines IL-1, TNF-α, and IL-6, and increased levels of IFN-γ, promoting the host immune response to contain infection successfully.20
NK cells also play an important role in the innate immune defence and control of M. tuberculosis infection through cytolytic activity.26 Like macrophages, NK cell functions are also impaired with low levels of GSH, allowing improved intracellular survival of M. tuberculosis.26 The authors observed that treatment of NK cells with a combination of IL-2, IL-12, and NAC caused a significant increase in NK cell cytolytic activity and, therefore, control of M. tuberculosis infection. These growth inhibitory effects on M. tuberculosis correlated with an increased expression of the Fas ligand and CD40 ligand on the cell surface of NK cells.22 With these findings, there is strong evidence that NK cells and macrophages work together to combat intracellular M. tuberculosis infection, and GSH has been shown to enhance this.26
GSH does not only support the innate immune response in controlling M. tuberculosis infection but study results have revealed that it is also important to the function of T cells of the adaptive immune response. A study of T cells derived from HIV+ individuals showed that the cells were deficient in GSH, as well as having decreased levels of Th1 cytokines, leading to increased growth of M. tuberculosis inside the host’s macrophages.27 When GSH levels were enhanced, T cells were able to inhibit the growth of M. tuberculosis inside the macrophages and also produced increased levels of IL-2, IL-12, and IFN-γ, essential for the Th1 response and control of intracellular pathogens.27
In addition, the authors performed a double- blind study in which HIV+ individuals with CD4+ T cell counts <350 cells/mm3 received either a placebo empty liposomal supplement or liposomal-GSH (L-GSH) (ReadiSorb™, Your Energy Systems, Palo Alto, California, USA) for 3 months. Prior to supplementation, baseline measurements of the HIV+ subjects demonstrated low levels of IL-2, IL-12, and IFN-γ, and high levels of IL-6, IL-10, TGF-β, and free radicals compared to the healthy subjects. Following the 3-month supplementation of L-GSH, there was an increase in GSH, as well as IL-2, IL-12, and IFN-γ, and a decrease in IL-6, IL-10, and free radicals, with stabilisation in the levels of IL-1, IL-17, and TGF-β compared to the placebo group, who showed no significant differences.28 Overall, L-GSH supplementation provided restoration of redox homeostasis and cytokine balance, thereby aiding the immune system in the control of infections.
Furthermore, it has been demonstrated that higher concentrations of GSH help to keep HIV-1 in its latent state.29 The GSH-synthesis inhibitor buthionine sulfoximine has been used in combination with histone deacetylase inhibitors as a complementary strategy to induce HIV-1 activation from quiescence by creating an oxidative environment that helps to stimulate HIV-1 transcription.29,30 Due to the critical role that redox signalling plays in the pathogenesis of HIV-1 and HIV–TB coinfection, genetically encoded redox-sensitive green fluorescent proteins (roGFP), particularly a roGFP-based specific bioprobe of GSH redox potential (EGSH; Grx1-roGFP2), have been created. This specific bioprobe allows for precise levels of EGSH to be measured, particularly levels that would suggest reactivation of HIV.31 In addition, it has been reported that bioactive lipids synthesised by drug-resistant strains of M. tuberculosis reactivate HIV-1 through increased intracellular EGSH, thus providing a more oxidative environment and suggesting that the cohabitation of these two pathogens allows for an enhanced environment for growth and pathogenesis.31
TRANSACTIVATOR OF TRANSCRIPTION PROTEIN
The transactivator of transcription (TAT) protein is a 101 amino acid protein coded by the HIV viral genome.32 It is a highly conserved regulatory protein of 14 kDa that operates within the infected host cell nucleus as an efficacious activator of HIV viral gene transcription.33 TAT plays a major role in HIV replication because it upregulates transcription from the viral long terminal repeat promoter by binding to the TAR hairpin in the nascent RNA transcript, preventing the host cell from abruptly stalling the transcriptional process.34 TAT functionality is vital for stable HIV replication due to the highly sensitive nature of the HIV promoter, which can be inhibited by various cellular regulators in the absence of TAT. The TAT- induced high rate of transcription of HIV allows the virus to outmatch the host immune response.
Furthermore, TAT directly participates in HIV infection due to its ability to act as an exotoxin, inducing cytotoxic and apoptotic effects on neighbouring T cells and resulting in high oxidative stress in the body.34 This action results in the depletion of GSH levels; low GSH levels are associated with poor survival among HIV patients.35 Moreover, TAT can increase TGF-β, causing a deficiency in the number of de novo synthesis enzymes, reducing GSH levels further.36 Ultimately, TAT allows increased virulence of HIV, inadvertently decreasing serum GSH levels, and can be mitigated by ingesting L-GSH as a potential therapeutic agent.
SUMMARY AND FUTURE DIRECTIONS
Compromised CD4+ T cell counts in HIV-infected individuals and AIDS patients predispose them to a 26–31-fold greater risk of developing TB, as well as other potentially fatal opportunistic infections.37 Currently, there is no cure for HIV and the present standard treatment for controlling HIV progression involves the use of antiretroviral therapy (ART) that targets different stages of the HIV lifecycle and enzymes essential for HIV replication and survival. Currently, 18.2 million people are receiving ART worldwide, but in the majority of patients, ART alone or in combination proves ineffective at preventing eventual progression of chronic HIV infection to AIDS or for the treatment of acute cases of AIDS.28 This is partially due to the high mutation rate of HIV and the rapid development of mutant HIV strains that are resistant to antiviral drugs. Furthermore, studies suggest that various ART combinations increase oxidative stress in HIV patients, contributing to a further redox imbalance in HIV-infected individuals.36,38 Modern-day HIV/AIDS research aims to formulate a vaccine or chemotherapeutic agent with the ability to completely ameliorate or circumvent HIV pathogenesis and infection. Until a cure is found, there is a need for other therapeutic agents and adjuncts to improve the prognosis of HIV patients and decrease complications due to opportunistic pathogens.
TB harbours a global burden, with one-third of the world’s population latently infected with M. tuberculosis.39 NAC is a GSH prodrug commonly used clinically for the treatment of pulmonary TB.40 In HIV patients coinfected with TB, HIV causes upregulation of TGF-β, suppressing GCL in the de novo pathway of GSH synthesis. L-GSH supplementation has been shown to provide increased immunological protection against HIV progression and resistance to opportunistic infections by restoring levels of GSH, correcting redox homeostasis, and downregulating the levels of TGF-β. In contrast to NAC treatment, L-GSH is able to bypass the de novo pathway, potentially acting as a more effective therapeutic agent in the treatment of concurrent HIV and TB infections. Extrapulmonary TB has also become increasingly common since the emergence of the HIV-1 infection, seen in >50% of patients with concurrent AIDS and TB. The most common and severe form of extrapulmonary TB is TB meningitis, causing pronounced morbidity and mortality.41 Recent research has reported decreased levels of GSH and increased levels of free radicals in brain tissue samples of HIV individuals, reducing neuroprotection and increasing the risk and occurrence of TB meningitis.41 Additionally, studies have reported that individuals with Type 2 diabetes mellitus (T2DM) have decreased GSH levels due to compromised GSH synthesis and metabolism enzymes. T2DM individuals are at a 2–3-fold greater risk of developing TB compared to those without T2DM.39
Research indicates that L-GSH shows promise as an effective adjunctive treatment in patients with HIV or TB, and studies have reported L-GSH may also play a potential role in the treatment of other human pathologies that cause a decrease n the levels of GSH, a redox imbalance, and increased levels of TGF-β. Future studies are needed to optimise the dosage of L-GSH supplementation in HIV+ individuals with CD4+ T cells counts <200 cells/mm3, as well as in individuals with CD4+ T cell counts between 200 and 350 cells/mm3, in order to improve HIVprognosis and control TB progression.28



