
Abstract
Ibuprofen is a nonsteroidal anti-inflammatory drug (NSAID) and is a widely used medication. One indication of NSAID use is long-term chemoprevention to decrease the risk of developing various types of cancer, in particular colorectal cancer. The molecular mechanism behind the antitumour properties of NSAID has been largely attributed to inhibition of the enzyme cyclooxygenase. In this review article, the authors highlight that additional mechanisms of NSAID, especially ibuprofen, action exist that are related to cell signalling and the modulation of gene expression, including alternative splicing. For example, the authors describe how ibuprofen inhibits expression of the tumour-related splicing variant RAC1b, which is overexpressed in a specific subset of colorectal tumours. The mechanism involves changes in the phosphorylation of splicing factors that regulate this alternative splicing event. According to recent studies, ibuprofen interferes with signal transmission via protein kinases, a process which is frequently altered in cancer cells.
INTRODUCTION
Ibuprofen belongs to the group of nonsteroidal anti-inflammatory drugs (NSAID) used to treat diverse inflammatory processes, pain, or fever. The mechanism underlying the effects of ibuprofen stems from the inhibition of cyclooxygenase (COX) activity, which is required for prostaglandin (PG) synthesis.1 PG are produced from plasma membrane-derived arachidonic acid and local PG production has hormone-like effects. Two COX isoforms are expressed in human tissues: the constitutively expressed COX-1 isoform exists in most tissues while the COX-2 isoform is strongly induced during the inflammatory response, including pathological conditions of chronic inflammation and colon cancer.2 Among different COX-2-derived products, the highest PGE2 levels are found in tumours and affect various processes, including cell proliferation and apoptosis.3 In normal physiology, PGE2 plays a role in the maintenance of the gastrointestinal mucosa regulating processes, such as mucus secretion and blood vessel dilation.4 Thus, prolonged NSAID treatment can lead to side effects, including intestinal bleeding. Most NSAID, including ibuprofen, inhibit both COX isoforms.
Prophylactic use of NSAID has been documented to reduce the risk of dying from colorectal cancer.5-9 For example, a 300 mg daily dose of aspirin over a period of 10 years revealed a statistically significant protective effect.7,10,11 A similar risk reduction was reported with a daily ibuprofen dose of 200 mg8,11-19 for various tumour types: 51% reduction in risk for colon, 72% for breast, 62% for prostate, and 59% for lung cancer.19
HOW DOES IBUPROFEN PREVENT CANCER?
Accumulating evidence has revealed that inflammation promotes tumourigenesis,20,21 in particular when the tissue is under chronic inflammatory conditions. Within the tumour microenvironment, inflammatory cells exchange signals with tumour cells. Stromal cells secrete survival factors for tumour cells while tumour cells produce cytokines, which trigger the proteolytic remodelling of the extracellular matrix by stromal cells, or the formation of new blood vessels.20,22 Ibuprofen inhibits COX activity and the subsequent generation of proinflammatory PG; this action is thought to underlie the chemopreventive effect of ibuprofen. PGE2, for example, activates G protein-coupled PGE2 receptors that stimulate various signalling pathways involved in cell proliferation and survival.23,24
In this article, the authors review additional mechanisms of action that are independent of COX-2 inhibition with the aim of increasing awareness that the clinical effects of ibuprofen can be mediated by several cellular processes. The presented evidence was retrieved from the PubMed search engine using “ibuprofen AND cancer” as the search term. Studies reporting COX-independent effects, including those conducted in the authors’ laboratory, were selected for review.
ADDITIONAL MECHANISMS THROUGH WHICH IBUPROFEN INHIBITS TUMOUR CELLS
In 2015, Matos and Jordan25 reviewed the treatment of cancer cells with ibuprofen. HCT-116 colorectal cells do not express COX-2, but the treatment with 2 mMol/L ibuprofen produced proapoptotic effects.26 Ibuprofen at a low concentration of 100 µMol was further identified as a direct and COX-independent ligand of peroxisome proliferator-activated receptor gamma (PPARγ),27 and was shown to stimulate its nuclear activity in rat models of colon cancer formation.28 Thus, the proapoptotic action observed for ibuprofen may in part result from PPARγ activation, which leads to the downregulation of the antiapoptotic transcription factor NFκB.28
Another COX-independent cellular response following ibuprofen treatment was reported to involve P75NTR, a member of the TNF receptor superfamily. Treatment of cancer cells with 1 mMol/L ibuprofen resulted in a p38 mitogen-activated protein kinase pathway-dependent stabilisation of p75NTR mRNA stability, leading to increased expression levels29 and induction of apoptosis and growth suppression.30
A similar apoptosis-promoting action was reported in HCT116 cells, when ibuprofen treatment (1.5 mM for 24 hours) was found to sensitise these cells against the TNF-related apoptosis-inducing ligand.31 The underlying mechanism involved expression of the membrane receptor for TNF-related apoptosis-inducing ligand: death receptor 5, another member of the TNF receptor superfamily.
Ibuprofen treatment (1 mMol/L for 24 hours) was further reported to significantly reduce the nuclear levels of β-catenin in SW480 and DLD-1 colorectal tumour cells. Consistently, the expression of one of its transcriptional targets, the pro-proliferative cyclin D1 gene, was suppressed.32 Although the underlying mechanism remains to be determined, this effect of ibuprofen seems of special interest for colorectal cancer prevention because excessive β-catenin signalling can cause inappropriate growth stimulation of colon mucosa stem cells.33
Concurrent to the effect on β-catenin signalling, ibuprofen also interfered directly with the NFκB pathway. A rapid effect of ibuprofen treatment observed in cells is the inhibitory phosphorylation of GSK-3β at serine 9.32 This modification was found to negatively regulate NFκB signalling, at a step downstream of the degradation of its inhibitor protein IkBα, and to suppress the expression of anti-apoptotic NFκB target genes, such as BCL2 and BIRC5.
Other examples for COX-independent effects of 100 µMol ibuprofen include the inhibition of integrin expression in neutrophils34 or the caspase-mediated release of proinflammatory cytokines in HCT-116 and HeLa cells.35
IBUPROFEN, ALTERNATIE SPLICING, AND CANCER
Cancer cells differ in their gene expression programme from their corresponding differentiated normal cells. Besides transcriptional regulation at gene promoters, the past 15 years have clearly revealed that alternative splicing serves as a significant mechanism for the regulation of gene expression. For example, alternative splicing generates transcript variants that can either be non-functional and rapidly degraded or be translated into protein isoforms with different, sometimes antagonistic, functional properties due to differential use of functional protein domains.36,37
Recently, inhibition of the alternative splicing variant RAC1b was identified as another COX-independent effect of ibuprofen.38 Colon inflammation was shown as one trigger for increased expression of the tumour-related RAC1b protein, a splice variant of the small GTPase RAC1. RAC1b protein contains an additional domain encoded by a 57 base pair-long alternative exon (exon 3b), which confers increased protein activation, generating a hyperactive variant able to stimulate NFκB signalling.39-42 When colorectal cells were treated with ibuprofen, but not with aspirin or flurbiprofen, both the mRNA and protein levels of RAC1b were markedly reduced in vitro and in vivo.38 Whereas many studies on the effect of NSAID on tumour cell viability used concentrations of up to 2 mMol/L,43 the effect of ibuprofen on alternative splicing of RAC1b was observed at low doses of 100 µMol. Interestingly, ibuprofen inhibited RAC1b-positive HT29 colorectal cells more than normal colonocytes and also affected their growth as subcutaneous tumour xenografts in mice. The inhibitory effect of ibuprofen could be rescued when a splicing-independent RAC1b cDNA sequence was expressed in HT29 cell.38 This suggests that ibuprofen acts directly on the alternative splicing event.
Another report on the modulation of alternative splicing was obtained when prostate cancer cells received combined treatment of ibuprofen and epigallocatechin-3-gallate (EGCG), a green tea component with anticarcinogenic properties that promotes G0/G1 cell cycle arrest and apoptosis. In this case, the balance between anti and proapoptotic splicing variants of BCL-X and MCL-1 was shifted towards the shorter and proapoptotic BCL-X(S) or MCL-1(S) variants.44 Although the mechanism was not fully identified, it involves activation of protein phosphatase PP1, which is known to dephosphorylate regulatory proteins involved in pre-mRNA splicing.
MECHANISM OF SPLICING MODULATION BY IBUPROFEN
When protein-coding genes are expressed in human cells, RNA polymerase 2 generates a primary transcript, the pre-mRNA, which contains coding exons separated by intronic sequences. While transcription is ongoing, conserved nucleotide sequences around each exon–intron junction are recognised by the spliceosome, a macromolecular machinery involving five small nuclear ribonucleoprotein particles (U1, U2, U4, U5, and U6 small nuclear ribonucleoprotein),45,46 which then removes introns during the process of mRNA splicing. The function of the spliceosome is assisted by splice enhancer or silencer elements, short sequences found in exons or introns, which either promote or inhibit productive recognition of a given exon by the spliceosome. Splicing factors recognise these splice enhancer or silencer elements, which mostly belong to the serine and arginine rich protein family or the heterogeneous nuclear ribonucleoproteins. They often act antagonistically, so that the modulation of binding provides a mechanism that allows inclusion or skipping of alternative exon and thus the generation of variant transcripts. Altogether, the set of splicing factors expressed in a given cell and their relative expression levels in the cell nucleus operate in a combinatorial mode to regulate alternative splicing.
In the case of RAC1b, alternative splicing is regulated by an enhancer element in exon 3b, which is recognised by the splicing factor SRSF1, and an adjacent silencer element recognised by SRSF3.47
In human colorectal cells, the availability of SRSF1 in the nucleus is the main factor regulating inclusion or skipping of exon 3b.48
One mechanism through which ibuprofen does affect alternative splicing in cells is the phosphorylation status of SRSF1. Cell fractionation and immunoblot experiments revealed that ibuprofen treatment caused a reduction in SRSF1 phosphorylation (unpublished data). By contrast, aspirin treatment had no such effect on SRSF1. This showed that the inhibitory effect of ibuprofen on RAC1b splicing involved post-translational regulation of SRSF1 subcellular localisation.48
The main protein kinase responsible for SRSF1 phosphorylation is SRPK1, which is found both in the cytoplasm and in the cell nucleus.49,50 This process is, in part, controlled by growth factor receptor signalling.51 As shown and described in Figure 1, the authors observed that ibuprofen treatment induced translocation of SRPK1 from the nucleus into the cytoplasm, and this correlated with reduced levels of SRSF1 phosphorylation and RAC1b protein as detected in whole cell lysates by western blot. No such effect was observed when cells were treated with aspirin under the same conditions, underlining the COX-independent action of ibuprofen and the specificity of its effect on splicing factor modulation.
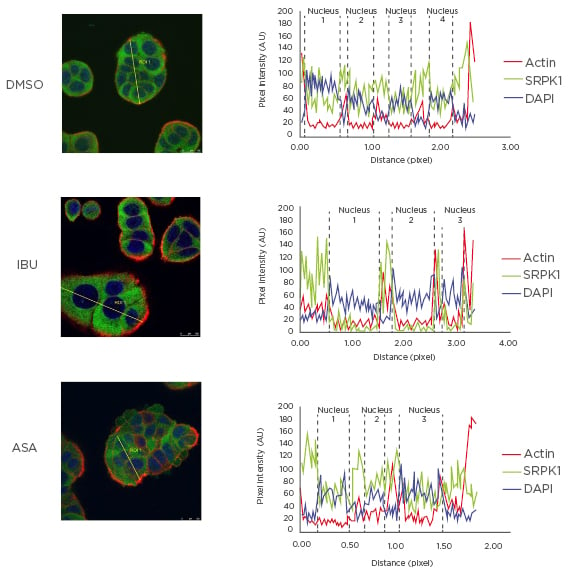
Figure 1: Effect of ibuprofen on subcellular localisation of protein kinase SRPK1.
HT29 colorectal cells were incubated for 48 hours with either a DMSO control solvent, 500 µM ibuprofen, or 500 µM aspirin and then fixed for immunofluorescence microscopy. Shown is the coloured overlay of three confocal immunofluorescence images (left), which detected cell nuclei in blue (DAPI), the localisation of endogenous SRPK1 protein in green (anti-SRPK1, BD Biosciences, San Jose, California, USA), and the actin cytoskeleton in red (Phalloidin-Texas Red). The nucleus and cytoplasm distribution of the three fluorescent signals was analysed along optical sections (yellow lines) across several cells by plotting pixel intensities along the traced path (right graphs). In control and aspirin-treated cells, SRPK1 signals (green) were localised both to the cytosol and the cell nucleus (blue); however, in ibuprofen-treated cells, nuclear signals for SRPK1 are nearly absent.
AU: arbitrary units; ASA: aspirin; DAPI: 4′,6-diamidino-2-phenylindole; DMSO: dimethyl sulfoxide; IBU: ibuprofen; SRPK1: serine/threonine-protein kinase.
Another mechanism through which ibuprofen can regulate splicing is the transcriptional modulation of splicing factor-encoding genes. The splicing factor SRSF3, for example, was previously described to be a direct transcriptional target for β-catenin/TCF signalling and ibuprofen has been found to downregulate β-catenin/TCF signalling in colorectal cells.52 A reduction in SRSF3 transcription and the consequent decrease in its nuclear levels will affect a variety of splicing variants.
Further research may unravel that, besides SRSF1 and SRSF3, other splicing factors are also modulated by ibuprofen treatment, either by regulation of their expression levels, their subcellular localisation, or their RNA-binding activity. These effects will most likely also include COX-dependent mechanisms as many of the PGE2 stimulated pathways23,24 have been described to affect alternative splicing regulation.53 It can thus be expected that ibuprofen treatment will affect a larger set of alternative splicing events in cancer cells and that these contribute to the described antiproliferative and proapoptotic effects.
CONCLUSION
Although ibuprofen has been used for chemopreventive therapies against cancers in the gastrointestinal tract, our understanding of the molecular mechanisms underlying the antineoplastic activity of ibuprofen is still rudimentary. Recently described cellular pathways linking ibuprofen to the COX-independent modulation of alternative splicing are summarised in Figure 2.
A better characterisation of its target molecules and their signalling pathways may provide opportunities for precision medicine approaches in cancer therapy or chemoprevention regimens. For example, the inhibitory effect on alternative splicing of RAC1b may benefit a subgroup of colorectal cancer patients characterised by serrated polyp morphology, BRAF mutation, and RAC1b overexpression. However, the deregulation of splicing factor SRSF1, which was described in this case, is most likely only the tip of the iceberg. It is now known that deregulation of splicing factors will inevitably affect a network of alternative splicing changes and this can be expected to have significant impact on cancer cell biology. A more systematic study with genome-wide determination of transcriptome changes should clarify the therapeutic opportunities that may arise from the COX-independent action of ibuprofen.
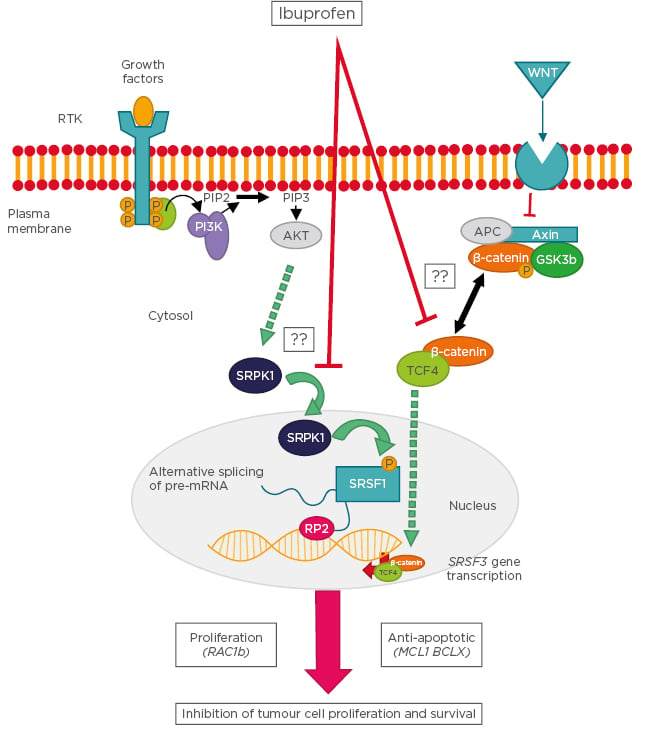
Figure 2: Schematic representation of the cellular pathways linking ibuprofen to the cyclooxygenase-independent modulation of alternative splicing.
Following stimulation of receptor tyrosine kinases, the PI3K becomes activated and leads to phosphorylation of SRPK1, which enters the nucleus and phosphorylates the splicing factor SRSF1. SRSF1 binds to specific recognition motifs on nascent pre-mRNA transcribed by RNA polymerase 2, thus affecting alternative splicing decisions. In a parallel pathway, Wnt ligands stimulate their plasma membrane receptor leading to inhibition of the β-catenin destruction complex and accumulation of a cytosolic β-catenin/TCF4 complex. This complex enters the nucleus and binds to gene promoters, including that of splicing factor SRSF3, the expression levels of which determine the outcome of specific splicing events. Examples of splicing variants affected by these pathways are RAC1b, MCL1, and BCLX. Question marks indicate that the molecular mechanism is still unknown.
AKT: protein kinase B; APC: adenomatous polyposis coli; GSK: glycogen synthase kinase; MCL1: myeloid Cell Leukemia Sequence 1; PIP2: phosphatidylinositol 4,5-bisphosphate; PIP3: phosphatidylinositol (3,4,5)-trisphosphate; PI3K: phosphoinositide 3-kinases; RAC1: Ras-related C3 botulinum toxin substrate 1: RP2: RNA polymerase 2; RTK: receptor tyrosine kinases; SRPK1: serine/threonine-protein kinase; SRSF: serine and arginine rich splicing factor; TCF4: transcription factor 4; Wnt: wingless/integrated.






