Abstract
It has been stated that developing a drug that can attack mutated RAS proteins is ‘the Holy Grail’ of cancer therapeutics. Through a series of unexpected findings, the authors discovered that the irreversible epidermal growth factor receptor 1/2/4 inhibitor neratinib (HKI-272, Nerlynx®) was not only an inhibitor of those receptor tyrosine kinases, but could additionally cause receptor internalisation and degradation. To the author’s surprise, the negative control receptors c-MET and c-KIT were also degraded after neratinib exposure, albeit with a slower time-course. This appeared to be attributable to neratinib attacking receptor tyrosine kinases localised in quaternary structures. It was reasoned that neratinib had the potential to downregulate the expression of other plasma membrane localised signalling proteins, particularly RAS. In a variety of tumour types, neratinib could reduce the expression of wild type (Kirsten) and mutant (Neuroblastoma) RAS (K-RAS/N-RAS, respectively). It was subsequently demonstrated that mutant Gα proteins in uveal melanoma could also have their expression reduced by neratinib. Neratinib was shown to be an inhibitor of sterile 20 serine/threonine kinases. Acting as an inhibitor of sterile 20 serine/threonine kinases, combined with RAS inhibition, neratinib enhanced the phosphorylation and degradation of the Hippo pathway effectors yes-associated protein and transcriptional coactivator with PDZ-binding motif. In malignancies expressing a mutant K-RAS, yes-associated protein and transcriptional coactivator with PDZ-binding motif are localised in the nucleus where they cooperate with mutant K-RAS signalling to promote growth, invasion, and chemotherapy resistance. Thus, whilst neratinib is not a direct inhibitor of mutant RAS signalling, the Holy Grail, it nonetheless represents, as did the beacon atop Castle Anthrax, at least something ‘Grail-shaped’.
WE ARE THE SCIENTISTS WHO SAY NERATINIB
The drug neratinib (HKI-272, Nerlynx®) was originally developed by Wyeth Research Laboratories, with its initial characterisation published in 2004.1 Several years later, as part of a comprehensive screening study examining the inhibitory properties of 40 kinase inhibitors against approximately 400 kinases, neratinib was shown, using computational chemical biology techniques, to be a potent inhibitor. This was not only of the receptor tyrosine
kinases epidermal growth factor receptor 1/2/4 (ERBB1/2/4), but also of multiple sterile 20 (Ste20) serine/threonine kinases.2 In the case of MAP4K5, neratinib was claimed to be a more potent inhibitor of this kinase compared to ERBB1. In the case of mammalian Ste20-like protein kinase 3 (MST3) and MST4, neratinib had a similar efficacy for their inhibition as the drug did for ERBB2 (Figure 1). Despite this surprising observation, no published studies have yet explored this possible ‘off-target’ biology surrounding the actions of neratinib. Subsequently, neratinib received U.S. Food and Drug Administration (FDA) approval as a neoadjuvant therapy in HER2+ breast cancer.3
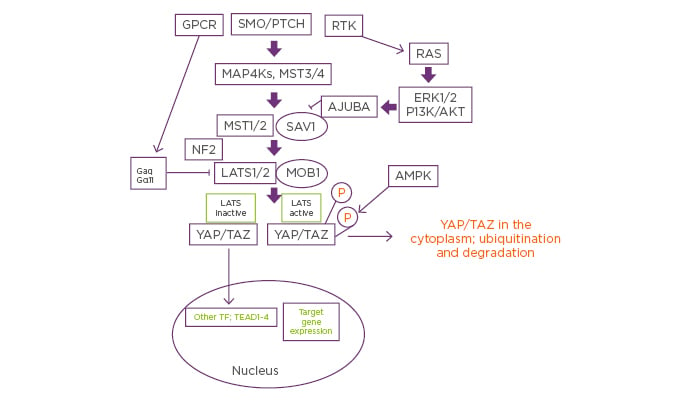
Figure 1: Putative mechanisms by which neratinib could coordinately control mutant RAS and Hippo Pathway signaling.
Signals from plasma membrane receptors can either stimulate or inhibit Hippo pathway functionality. Classic Hippo pathway signalling proceeds from Smoothened and Patched and via the regulation of MAP4K results in the phosphorylation and activation of MST1/2. Activated MST1/2 phosphorylate and activate LATS1/2. Activated LATS1/2 phosphorylate YAP and TAZ, which causes cytoplasmic sequestration of YAP and TAZ, prior to their eventual degradation. Through a variety of mechanisms, the activities of MST1/2 and LATS1/2 can be reduced via crosstalk from other signaling pathways. Thus, mutant RAS signaling, via ERK and AKT signaling and a redistribution of chaperoning/complex effectors, can block MST1/2 signaling. Mutant Gα proteins, as found in uveal melanoma, can act to prevent LATS1/2 activation. Reduced MST/LATS activities result in YAP/TAZ dephosphorylation, permitting these co-transcription factors to interact with TEAD proteins to enhance the expression of proteins that promote growth, invasion and chemotherapy resistance. Neratinib can block the actions of mutant RAS as well as of mutant Gα proteins. Furthermore, it activates the AMPK, all of which would be predicted to prevent YAP/TAZ and TEAD proteins colocalising in the nucleus where they facilitate tumourigenic cellular behaviour.
AKT: protein kinase B; AMPK: 5′ AMP-activated protein kinase; ERK: extracellular regulated kinase; GPCR: G-protein-coupled receptors; LATS: large tumour suppressor kinase; MOB: MOB kinase activator; MST: mammalian Ste20-like protein kinase; NF2: neurofibromatosis type 2; P: phosphorylated; PTCH: Patched; RTK: receptor tyrosine kinase; SAV1: salvador homolog 1; SMO: Smoothened; TAZ: transcriptional coactivator with PDZ-binding motif; TEAD: TEA domain family member 1; TF: transcription factor; YAP: yes-associated protein.
The authors’ initial interest in neratinib was based upon data demonstrating that ERBB1/2/4 inhibitors, particularly the irreversible inhibitor afatinib, could combine with the JAK1/2 inhibitor ruxolitinib to kill tumour cells.4 Afatinib is approved as a non-small cell lung cancer (NSCLC) therapeutic, and in another project, multiple independent afatinib-resistant NSCLC clones had been generated from in vivo exposure of established H1975 tumours that express ERBB1 L858R/T790M.5 Characterisation of these cells revealed no additional adaptive mutations in cancer hot-spot genes, and instead, these cells had reduced their expression of the tumour suppressor phosphatase and tensin homolog (PTEN) and increased expression of the PTEN-regulatory E3 ligase NEDD4.6 Neratinib and afatinib were both capable of killing parental H1975 clones, but only neratinib killed the afatinib-resistant H1975 clones; additionally, the resistant cells were significantly more sensitive to neratinib even though they expressed less of the proposed primary neratinib targets ERBB1/2/4. These findings implied that neratinib “had to be inhibiting something else” so that it could kill the afatinib-resistant NSCLC cells.
RAS: THE ONCOGENE SUPREME
Very few modern targeted cancer therapeutics have significant clinical activity when used as a stand-alone medication. In general, where single agent drugs have shown activity, the tumour cells exhibited an exquisite addiction to signals emanating from one particular mutated enzyme, e.g., BCR-ABL; ERBB1 L858R.7 One family of oncogenes that are rational single agent targets, but that have proven very difficult to block, are those belonging to the RAS family.8-10 RAS proteins are small GTPases that regulate cellular signalling cascades downstream of receptor tyrosine kinases to control cell growth, proliferation, and differentiation.11-14 The three RAS isoforms H (Harvey), N (Neuroblastoma), and K (Kirsten)-RAS are expressed in mammalian cells; for example, K-RAS is mutated in 90% of pancreatic tumours.15 In mutated RAS proteins, the GTPase activity of the protein is greatly reduced, and thus the RAS protein is permanently capable of activating downstream effectors such as RAF-1.16 To act as signal transducers, RAS proteins must also be localised to the inner leaflet of the plasma membrane by a COOH-terminal membrane lipid anchor. For K-RAS, the anchor comprises a covalently attached COOH-terminal cysteine farnesyl-methyl ester operating together with a polybasic motif of 6 lysine residues that provide electrostatic membrane affinity. Clinically relevant β-hydroxy β-methylglutaryl-CoA reductase inhibitors, i.e., statins, reduce the levels of farnesyl substrate and reduce the amount of K-RAS that can localise in the plasma membrane.17
Although for nearly 40 years oncogenic forms of RAS have been recognised as key drivers of cancer growth, invasion, and chemotherapy resistance, they have also been considered as ‘undruggable’.18-20 Hence, therapeutic inhibition of mutant RAS signaling came to be considered as the ‘Holy Grail’ in the field of cancer therapeutics.21 In recent years, attempts have been made to develop agents that directly inhibit mutant RAS function, but these inhibitors at present only target a small fraction of RAS mutations.22-24 Initial studies with neratinib demonstrated that it not only blocked the kinase activities of ERBB family receptors, but that it also caused their degradation.25 In agreement with the concept that receptors reside in quaternary structures in the plasma membrane, neratinib also causes the degradation of c-MET and c-KIT that do not bind the drug. Based on these findings, it was reasoned that other signal transducing proteins, i.e., RAS isoforms, may also become degraded after neratinib exposure. Clinically relevant concentrations of neratinib within 4 hours reduced the expression of mutant K-RAS proteins in pancreatic cancer cells by 30%; this down-regulation only resulted in 15–20% tumour cell death after 24 hours.
RAS: ADDITIONAL OPPORTUNITIES TO ATTACK ITS ONCOGENIC CAPABILITY
In the case of neratinib and mutant K-RAS, two additional concepts were then developed to enhance the downregulation effect and to increase the killing. The combination of neratinib with histone deacetylase (HDAC) inhibitors, including sodium valproate, entinostat, panobinostat, and vorinostat, resulted in a rapid 50–70% reduction in K-RAS expression associated with >40% of the cells dying within 24 hours.26,27 The second concept involved directly attacking the mutant K-RAS protein itself through mechanisms independent of those induced by neratinib. K-RAS is phosphorylated by protein kinase G, a downstream target of phosphodiesterase 5 inhibitors such as sildenafil (Viagra®), which leads to K-RAS leaving the plasma membrane.28 For K-RAS to be localised in the plasma membrane it must be prenylated, and inhibitors of β-hydroxy β-methylglutaryl-CoA reductase, including statins such as atorvastatin (Lipitor®), reduce farnesyl substrate levels required for the prenylation of K-RAS.29 Neratinib, sildenafil, and atorvastatin interacted in a greater than additive fashion to reduce K-RAS protein levels and to kill pancreatic cancer cells (Figure 2).27 One observation from preclinical in vivo studies in triple-negative breast cancer cells was that tumours previously exposed to neratinib and the HDAC inhibitor sodium valproate had permanently reduced their expression of ERBB1, K-RAS, and N-RAS 14 days after the final treatment.30 Thus, this drug combination appeared to have evolved the surviving tumour cells in vivo to have a less aggressive phenotype. A Phase I trial recently opened at Massey Cancer Center combining neratinib with the HDAC inhibitor valproate,31 with planned expansion cohorts at the recommended Phase II dose in patients carrying K-RAS-mutant tumours. One pancreatic adenocarcinoma patient, in the first cohort, has exhibited stable disease.
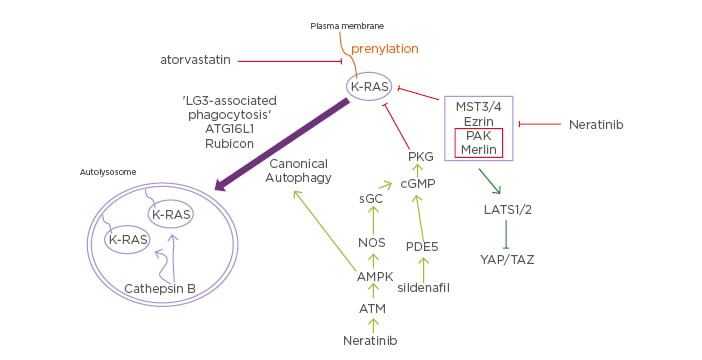
Figure 2: Putative mechanisms by which neratinib downregulates mutant Kirsten-RAS expression and inactivates Hippo pathway function.
Neratinib via inhibition of MST3/4 causes Rubicon-dependent phagocytosis and in parallel, neratinib activates ATM-AMPK signaling that causes autophagosome formation. Collectively, this results in the cathepsin-dependent degradation of mutant K-RAS. Atorvastatin via reduced prenylation and sildenafil via increased PKG-dependent phosphorylation of K-RAS also independently act to lower mutant K-RAS levels. As part of this process at the plasma membrane, neratinib reduces PAK1 phosphorylation that leads to dephosphorylation of the PAK1 substrate Merlin/ NF2. Dephosphorylated Merlin facilitates activation of LATS1/2, and active LATS1/2 phosphorylate YAP and TAZ. Phosphorylated YAP and TAZ leave the nucleus, preventing them from acting as transcriptional coactivators.
AMPK: 5′ AMP-activated protein kinase; ATG16L1: autophagy related 16 like 1; ATM: Serine-protein kinase ATM; cGMP: cyclic guanosine monophosphate; K: Kirsten; LATS: large tumour suppressor kinase; LC3: microtubule-associated proteins 1A/1B light chain 3B; MST: mammalian Ste20-like protein kinase 3; NOS: nitric oxide synthases; PAK: Serine/ threonine-protein kinase, PDE5: phosphodiesterase type 5 inhibitor; PKG: protein kinase G; sGC: soluble guanylate cyclase; TAZ: transcriptional coactivator with PDZ-binding motif; YAP: yes-associated protein.
NOT THE COMFY CHAIR: NERATINIB IS A MULTI-KINASE INHIBITOR
As mentioned in the first paragraph, in addition to ERBB1/2/4, neratinib could potentially inhibit MAP4K serine/threonine kinases. These alternate/secondary neratinib MAP4K targets are not only expressed in epithelial carcinoma cells but also in haematopoietic tumour cells. This raised the possibility that neratinib, alone or combined with HDAC inhibitors, could be repurposed as an antileukemia or an antilymphoma drug. Not only did neratinib and HDAC inhibitors interact to kill acute promyelocytic leukaemia, acute myeloid leukaemia, and T cell lymphoma cells, the drugs interacted, in the absence of any ERBB family receptor expression, to reduce the protein levels of K-RAS and N-RAS in these cells. These findings suggested that specific inhibitors of MAP4K/Ste20 kinases, more potent and efficacious than neratinib, may have a wide utility as cancer therapeutics.
Canonical Hippo pathway signalling initially involves the MAP4K family enzymes MST1 and MST2 phosphorylating the intermediate kinases large tumour suppressor kinase (LATS) 1/2 and the docking protein/chaperone MOB1. LATS1/2 phosphorylate the yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) effector co-transcription factors.32-40 Phosphorylated YAP and TAZ are located in the cytosol whereas dephosphorylated YAP/TAZ are nuclear and regulate transcription. Phosphorylated YAP/TAZ in the cytoplasm are degraded via ubiquitination (Figure 1). In malignancies expressing a mutant K-RAS, YAP and TAZ are predominantly localised in the nucleus where they cooperate with mutant K-RAS signalling to promote growth, invasion, and chemotherapy resistance.38,39 High expression levels of YAP are clinically associated with greater metastatic spread of pancreatic cancer cells,39 and downstream of mutant K-RAS, the transcription regulator YAP is essential for neoplastic progression to pancreatic ductal adenocarcinoma.40,41 Well-described proteins whose expression is regulated by the Hippo pathway include cyclin E, cell-division cycle protein 20, solute carrier family 7 member 5, anillin, stathmin 1, SH2 domain-binding protein 1, N-myc downstream-regulated gene 1, collagen Type IV alpha-3, fascin, hyaluronan-mediated motility receptor, moesin, cytochrome P450, shisa family member 4, growth differentiation factor 15, and damage specific DNA binding protein 2.41-47 Cell-division cycle protein 20 overexpression in pancreatic cancer predicts for poor patient survival. Overexpression of the L-amino acid transporter solute carrier family 7 member 5 predicts for poor pancreatic patient survival. Elevated levels of the cytoskeletal protein stathmin 1 is associated with poorer patient survival. Transforming growth factor beta family member growth differentiation factor 15 is overexpressed in pancreatic cancer and has been proposed as a better biomarker for pancreatic cancer than CA19-9. N-Myc downstream regulated 1 is a key mammalian target of rapamycin (mTOR) complex 2 effector that regulates the stability of methyltransferases and alkylating agent resistance.
Neratinib reduced the phosphorylation of MST1, MST3, and MST4, yet increased the phosphorylation of LATS1/2. LATS1/2 activation correlated with enhanced phosphorylation and degradation of YAP and TAZ, an effect that was further increased when it was combined with an HDAC inhibitor (Figure 1).27 This implied that neratinib may cause a compensatory activation of an unknown MAP4K that can act to phosphorylate LATS1/2. The authors also determined whether neratinib, atorvastatin, and sildenafil could interact to alter YAP phosphorylation. Both atorvastatin and neratinib enhanced YAP phosphorylation, though neratinib more effectively suppressed K-RAS expression. The drugs interacted to further elevate YAP phosphorylation and to reduce K-RAS expression. Sildenafil enhanced the ability of neratinib plus atorvastatin to further suppress K-RAS expression and to increase YAP phosphorylation. Thus, the three agents in coordination acted to simultaneously block RAS and YAP function which was associated with significant amounts of tumour cell killing (Figure 2).
AUTOPHAGY: EVOLUTIONARY RECYCLING AND A CANCER CELL’S ACHILLES HEEL
Autophagy is an evolutionary conserved process found in single cell yeasts and in multicellular mammals.48,49 The basic role of autophagy is to recycle cellular components if they are damaged or denatured, or during times of nutrient stress, to maintain homeostasis and cell viability.50 The autophagosome initially forms around the damaged organelles and/or proteins, and the fuses with acidic endosomes to form an autolysosome.51 The organelles and proteins are subsequently degraded in the autolysosome and the degraded materials returned to the cell for new uses.52 The regulation of autophagy in mammalian cells can be simplistically viewed as alterations in signalling by mTOR that regulates the expression and phosphorylation of multiple proteins who control autophagosome formation, autophagosome fusion with endosomes, and autolysosome acidification.53-57 Simplistically, mTOR and the AMP-dependent protein kinase (AMPK) coordinately regulate the activity of the Unc-51 like autophagy activating kinase (ULK1). Phosphorylation of ULK1 at COOH-terminal sites by mTOR inactivates ULK1, for example at S757. Phosphorylation of ULK1 at sites closer to the NH2-terminus of the protein by the AMPK activates ULK1, for example at S317. Furthermore, it should be noted that signalling by the AMPK can itself cause inactivation of mTOR via phosphorylation of raptor; mTOR activity is generally thought to be maintained by upstream signalling from the PI3K/PTEN/protein kinase B pathway. This dynamic multi-site phosphorylation of ULK1 means that a cell can exquisitely control the ability of ULK1 to phosphorylate its key target: autophagy-related protein 13. Phosphorylation of autophagy-related protein 13 at serine 318 represents the key gate-keeper step for autophagosome formation. Important additional proteins, such as Beclin1 and autophagy related 5, also play essential roles in the formation of the double-membrane autophagosome. Under normal biological circumstances, the autophagosome matures and then fuses with endosomes that acidify, facilitating the proteolytic degradation of their contents, for example by cathepsin and calpain proteases.58-61 This transition process is termed ‘autophagic flux’.
Neratinib rapidly promoted the formation of autophagosomes, which was attributable to both activation of an ATM–AMPK–ULK1 pathway and by reduced protein kinase B and mTOR signaling.25-27 This autophagy effect was significantly enhanced by combined exposure of neratinib with HDAC inhibitors. Because the authors were using HDAC inhibitors, control studies were performed to examine the total expression of the various HDAC proteins, 1–11, in the tumour cells. The authors discovered that the combination of neratinib with HDAC inhibitors caused the protein expression of multiple HDAC proteins to rapidly decline, particularly HDAC1/2/3/6, an effect that was blocked by preventing autophagosome formation. For example, the expression of cytosolic HDAC6 and the chaperone it regulates, HSP90, were rapidly reduced via this process. The loss of HDAC6/HSP90 function has been shown by many laboratories to be highly detrimental to tumour cell growth and the maintenance of drug-resistance.62 As HDAC6/HSP90 function declines, the amount of unfolded protein within the cytoplasm and the endoplasmic reticulum increases, leading to prolonged intense PKR-like endoplasmic reticulum kinase-eIF2α endoplasmic reticulum stress signalling, and a dramatic reduction in the levels of protective proteins with short half-lives such as myeloid leukaemia cell differentiation protein-1.25,63,64
HDAC INHIBITORS AS ENHANCERS OF IMMUNOTHERAPY EFFICACY
HDAC inhibitors have generated interest within the checkpoint immunotherapy field where it has been argued that epigenetic modulation of protein expression by this family of drugs predisposes tumours to be more responsive to immunotherapeutic checkpoint inhibitory antibodies.65-67 In prior studies from this laboratory, using drug combinations such as neratinib plus valproate but also with combinations that lack an HDAC inhibitor (e.g., pemetrexed plus sildenafil), it was observed that the drug combinations simultaneously rapidly enhanced, again in an autophagy-dependent fashion, tumour cell expression of Class I major-histocompatibility, and decreased expression of programmed death ligand 1, ornithine decarboxylase, and indoleamine 2, 3-dioxygenase 1.25,63,66-68 Because the drug combination was reducing the levels of HDAC proteins via autophagy whilst simultaneously altering the expression of immunologically important proteins, the authors hypothesised that these two events were linked. Using molecular tools to knock down the levels of HDAC 1/2/3/10, alone or in combination, the authors recapitulated the effects on immunological protein expression that were observed following drug combination exposure. Thus, for drug combinations lacking any epigenetic modulator, to cause these changes in protein expression, required autophagosome formation; i.e., the drug-combinations reduced the levels of HDAC and HDAC expression was preserved when drug-induced autophagosome formation was blocked. These findings imply that any drug combination which causes prolonged endoplasmic reticulum stress signalling together with strongly enhancing autophagosome formation have the potential, via down-regulation of HDAC proteins themselves, to both cause tumour cell death in parallel with altering epigenetics/transcription and protein expression leading to enhanced tumour cell immuno-sensitivity.
For neratinib plus valproate, beyond showing that the two drugs interacted to suppress tumour growth, additional studies using checkpoint inhibitory immunotherapy antibodies were performed.25 Under controlled antibody conditions, a 3-day exposure of tumours to neratinib plus valproate, 13 days later, still resulted in significant reductions in the expression of K-RAS and ERBB1. The expression of several HDAC proteins, such as HDAC6, was also permanently reduced. These findings suggested that at least for this particular drug combination the expression of some oncogenes can be ‘reset’ to basal levels. Prior exposure of tumours to neratinib plus valproate enhanced the antitumour efficacy of both an anti-programmed cell death protein 1 antibody and an anti-cytotoxic T-lymphocyte-associated protein 4 antibody. These events were associated with immune cell infiltration into the tumour: M1 macrophages, activated natural killer cells, and CD8 T cells, all of which correlate with the observed antitumour response.
ADDITIONAL THOUGHTS: YOU’VE GOT TO THINK FOR YOURSELVES!
From a translational cancer research perspective, and with respect to the data discussed in this overview, it is also important to consider experimental design with small molecule therapeutic agents when measuring and assessing cellular responses such as viability, autophagosome formation, and the degradation of HDAC and chaperones. Many of the cell biology effects discussed in this manuscript, using clinically relevant drug concentrations of neratinib, HDAC inhibitors, statins, and PDE5 inhibitors, are modest, with alterations of phosphorylation or expression being within a 50% reduction or a 2-fold increase.25-27 Hence, it is possible that the in vitro data for neratinib, RAS, and YAP/TAZ using these physiologic concentrations may not induce enough of a biological alteration to actually kill tumours in a patient. On the other hand, using low clinically relevant drug concentrations for in vitro research is in stark contrast to the majority of cancer therapeutic manuscripts where much higher drug levels are used in vitro to observe effects with greater amplitudes, for example as described by Carrer et al.69 and the concentrations of statins used in their work. Thus, before any laboratory-based study is performed, it is vital for investigators to determine from the literature/Phase I trials the safe maximum plasma concentration and the area under the curve showing the plasma drug concentration over time. Usually, information is also provided by a drug company as to how much of their drug is protein-bound, probably inactive, and in the plasma. All of this information can be used to empirically judge at what concentration a drug should be used for cell-based studies, combined with other agents. For example, the maximum plasma concentration of sorafenib tosylate following a 400 mg ingestion is ~13 µM. However, sorafenib tosylate is >90% protein-bound in the plasma. Thus, for meaningful in vitro studies, the maximum drug concentration for cells growing in 10% (v/v) serum cannot realistically be >2 µM.70,71 The obvious reasoning for this approach is that studies using a drug at a physiologic concentration, such as neratinib at 100 nM or below, may yield different biological information on viability, autophagy, and cell signalling processes versus studies using concentrations at an order or two above the safely achievable plasma drug level in a patient.
CONCLUSION: THAT RABBIT’S DYNAMITE
In conclusion, neratinib and derivatives of this drug are potentially of great importance in the fight against mutant RAS cancers. Neratinib as a single agent can act in carcinoma cells to simultaneously downregulate ERBB family receptors, associated quaternary complex receptors, and mutant RAS proteins in parallel with it. As a result, YAP phosphorylation and translocation of the co-transcription factor to the cytoplasm, this suggests that additional combinations of neratinib with other yet to be defined agents could represent the end of the beginning of, in a rational on-target fashion, therapeutically controlling mutant RAS cancers.






