Abstract
The high frequency of allergic reactions (AR) to non-steroidal anti-inflammatory drugs (NSAID) has been linked to their extensive usage, resulting in adverse reactions that affect various systems and organs. This literature review, initiated by a case report of an immediate rash following NSAID administration for fever reduction, explores the possible mechanisms that drive such reactions. The results indicate that, in certain instances, NSAIDs can induce ARs specifically in the presence of an inflammatory febrile response following viral infections through a complex interaction of similar mechanisms. This intricate process involves the activation of proallergic-pyrogenic mediators like complement factor C5a, IL-1 β and IL-6, and TNF-α, in addition to the modulation of immune cells through specific signal pathways. Moreover, factors such as compromised skin barrier function in some severe skin ARs, and the activation of memory CD4+ T helper 2 cells by TNF-family molecules, contribute to the emergence of ARs in reaction to NSAID exposure during a febrile condition. This case report underscores the significance of thoroughly reviewing the patient’s medical history to ensure safe treatment. Meanwhile, the transient drug hypersensitivity reactions necessitate further exploration for a comprehensive understanding of the underlying mechanisms.
INTRODUCTION
Non-steroidal anti-inflammatory drugs (NSAID) are useful in treating musculoskeletal disorders, headaches, pain, fever, and others.1,2 Fever is a phylogenetically ancient host reaction to invading microorganisms and other noxious stimuli. Endothermic organisms produce febrile temperatures through endogenous heat production at the expenditure of a higher metabolic rate.3 Despite its advantage for host defence, most doctors, nurses, and patients use antipyretics to improve fever and well-being.
The widespread use explains the frequent allergic reactions (AR) to them, ranging from asthma and rhinitis to urticaria and angioedema, various skin eruptions, and anaphylactic shock.1,2 Drug allergy (DA) management represents a physician challenge and persistent distress for the patient. The disease history could establish the diagnosis of NSAID hypersensitivity, because often, skin prick tests with NSAIDs are not successful, and no reliable in vitro tests are available. The only determinative diagnostic test is the oral challenge, while a tolerance test serves us to identify alternative NSAIDs in a hypersensitised patient.1,4 Initiated by a case of repeated immediate NSAID-induced rash episodes when administered for fever release but symptom-free when taken for non-febrile events, the following review aimed to explore the common mechanisms involved in the allergic response to NSAIDs and fever.
A CASE DESCRIPTION
A 39-year-old subject admitted for respiratory febrile infection reported generalised urticarial rash events after intake of antipyretics (acetaminophen, ibuprofen, etc.) only during febrile episodes. The mentioned drug reaction occurred first after extensive combined therapy for chronic tonsillitis (first amoxiclav, then clarithromycin), and then after prolonged treatment for COVID-19 (ceftriaxone, levofloxacin, dexamethasone, etc.). The urticarial reaction occurred within the first hour after NSAID intake, and the emergent treatment with glucocorticoids resolved them within 5–6 hours without sequels. In more recent episodes, NSAID-related reactions happened before the beginning of any antibiotic therapy. In contrast, the further intake of any antipyretic drug didn’t induce allergic reactions when used for non-febrile events (such as headaches). The subsequent administration of any mentioned antibiotic after these episodes did not represent allergological clinical relevance.
The subject mentioned that previously an allergist considered the disease history impossible or inconsistent, and stated that a negative drug provocation test as a test of choice5,6 always excludes an AR. Being convinced about the presence of DA based on disease history, the authors commented for the patient that a negative result for the drug provocation test will be expected, and this does not necessarily predict an AR lack when NSAID should be used for fever reduction. In such circumstances, they considered aquatic baths at 29–33 °C and parenteral glucocorticoids the most suitable alternative against the fever. Appreciating the avoidance of suspicious drugs as the first choice, they suggested eventual premedication with antihistamines and leukotriene modifiers, and the controlled use of an NSAID only as add-on therapy (to large dose glucocorticoids) under medical surveillance.4,5,7 Cyclooxygenase (COX)-2 inhibitors (like rofecoxib) represent an additional therapeutical alternative, as studies in humans or animals conclusively confirm the pivotal role of COX-2 in the febrile response after natural episodes or exposure to pyrogens, respectively.8 However, this alternative may be unsafe since the subject is allergic to acetaminophen, an inhibitor of the inducible COX-2 subtype synthesised in the brain.
Regarding the differential diagnosis, negative serological results for Epstein-Barr virus, human herpesvirus 6, cytomegalovirus, etc., excluded a viral exanthema during the medical treatment of viral infection.6 Additional undetermined agents should be considered as a cause of such reactions. Especially in children, viruses may induce maculopapular rash or urticaria due to skin infiltration or immunologic response. In antibiotic-related skin eruptions, the culprit drug triggers the reaction after a transient virus-induced immune activation.9 Like in other studies, the skin test with acetaminophen and ibuprofen resulted negative, while basophil activation or lymphocyte transformation tests were unavailable.6,7,10 Further haematological and biochemical investigations did not differentiate between the two types of exanthema. Despite the difficulty distinguishing between infection-induced and drug-induced skin eruption in the acute phase, the outbreak of skin symptoms after the NSAID intake during different febrile infections supported a DA presence, including the cofactorial role of the infective agent in the transient reduction of immune tolerance. The urticaria outbreak before antibiotic exposure reinforced DA diagnosis and excluded the antibiotics’ hypersensitivity.
DISCUSSION
Biological Mechanisms of Fever
NSAIDs like acetaminophen and ibuprofen are widely used for antipyretic and analgesic purposes. They reach this effect due to COX inhibition, thereby impairing the ultimate transformation of arachidonic acid to prostaglandins (PG), prostacyclin, and thromboxanes.2 The crucial effect is the inhibition of PGE2, which binds to its receptors on thermoregulatory neurons in the anterior hypothalamus, primarily eliciting fever.11
Several studies demonstrated that febrile PGE2 synthesis implicates different biological factors. Concerning the immune cells, this effect involves lymphocytes, mononuclear phagocytes, or neutrophils that exacerbate the immune response.2,12,13 The experimental inoculation of common pyrogens, bacterial lipopolysaccharide, or polyinosinic:polycytidylic acid (poly I:C), induces an inflammatory response and immediate fever implying IL-1β, IL-6, TNF-α, anaphylatoxic complement factor C5a, granulocyte colony-stimulating factor, and platelet-activating factor.13-15 Another factor in the febrile response is the sympathetic stimulation of β3-agonist receptors in immune cells and the central nervous system.13,16
About the enzymes, the PGE2 synthesis needs COX-1 and COX-2, phospholipases A2, or terminal PGE synthases (PGES).14 The ‘inflammatory’ set comprises inducible COX-2 isoforms and microsomal PGES-1. The PGE2 receptors are multiple; one of them, EP3, is likely to be a primary ‘fever receptor’.14
The accelerated PGE synthesis and inflammatory rise of body temperature need the interaction between immune and neuronal factors. The early febrile response can occur through COX-independent mechanisms.11,17,18 In contrast, experimental suppression of some pyrogenic cytokines and sympathetic response can lead to the abrogation of PGE production and lack of febrile reaction.11,12,15,16 Apart from the generalised up-regulation, pyrogenic enzymes and cytokines are also synthesised or transported into certain central nervous structures, such as cerebral ventricles, endothelial and perivascular brain–blood barrier cells, microglia, hypothalamus, and cerebrospinal fluid.11,12,14,15,19 The PGE and other pyrogenic factors in the hypothalamus can activate a specific set of pathways, including the ventromedial preoptic area, which is a key regulatory site for thermoregulation, and the paraventricular nucleus, which produces autonomic and endocrine responses that cause hyperthermia.20-22 The ‘thermostat turn-up’ pathway activates the raphe pallidus, where premotor sympathetic neurons driving thermogenesis in the brown adipose tissue and skin vasoconstriction are located. Further, neuropeptides and peptide hormones modulate inflammatory signalling and thermo-effector pathways involved in fever.14,19-21
In summary, the febrile reaction implies an interaction between immune cells, endothelia, and certain thermoregulatory and effector formations in the central nervous system, which activate different inducible enzymes for the PGE2 synthesis (Table 1). Additional factors on the PGE2 stimulation include some sympathetic fibres and receptors, and many inflammatory pyrogenic factors like IL-1β, IL-6, TNF-α, or complement fraction C5a, which mediate the thermogenesis and hyperthermia. They act as necessary cofactors in the inflammatory acceleration of PGE2 synthesis due to COX-dependent and -independent mechanisms.
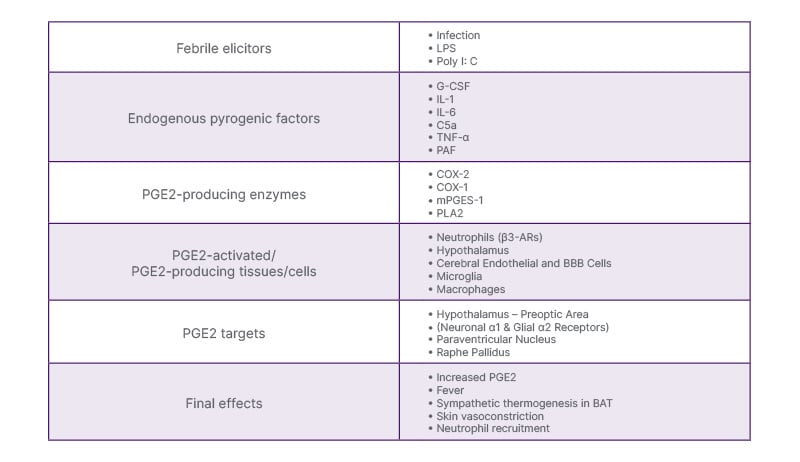
Table 1: Mechanisms of febrile response.2,11-22
β3-AR: adrenoreceptor β3; BAT: brown adipose tissue; BBB; brain–blood barrier;
C5a: complement factor C5a; COX: cyclooxygenase; G-CSF: granulocyte colony-stimulating
factor; LPS: bacterial lipopolysaccharide; mPGES-1: microsomal PGE synthase-1; PAF: platelet-activating
factor; poly I: C: polyinosinic: polycytidylic acid; PGE2: prostaglandin E2; PLA2: phospholipase A2.
Pyrogenic Factors Implicated in Immuno-Allergic Pathologies
Cytokines are highly inducible, secreted proteins mediating intercellular communication in the nervous and immune systems.17 Besides the infection-related events such as febrile reaction, inflammatory pain, or polymorphonuclear cell mobilisation,11,12-14,23-25 some immune-mediated components can exacerbate cellular responses and create complex pathways that lead to various clinical manifestations.2,13 So, pyrogenic compounds IL-1, IL-6, C5a, or TNF-α are involved in autoimmune and hypersensitive pathologies.26-31 NSAID-related DAs include drug reaction with eosinophilia and systemic symptoms (DRESS), toxic epidermal necrolysis (TEN), Stevens-Johnson syndrome (SJS), and T cell-mediated, IgE-mediated, and cross-hypersensitivity allergies, often mediated by above-mentioned acute phase reactants.7,27,29,32-36
In these cases, the pyrogenic cytokines activate many immune cells and complex processes. Thus, the mast cells (MC) play a crucial role in the baseline inflammatory processes related to IgE response or COX-1 inhibition after exposure to NSAIDs.37 During degranulation, MCs release preformed and de novo-formed inflammatory mediators like TNF-α or platelet-activating factor, mediating the increase of vascular permeability. In turn, MC activation occurs under the effect of C5a, or other inflammatory cytokines.27,28,38,39 Being produced at the early stage of allergen sensitisation, then TNF-α continues to promote the inflammation cascade in the effector phase of ARs.31 MCs, the most important effector cells in IgE-mediated hypersensitivity reactions, effectively combat bacterial infections by releasing antimicrobial peptides.40 Like the MCs, the cutaneous dendritic cells (DC) play a crucial role in allergen detection and processing, which leads to allergy development. Still, they are also essential in host defence against bacteria by releasing cytokines such as TNF and IL-17.40 Another source of proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α, are alternatively-activated macrophages (M2 macrophages), which, among others, exacerbate the inflammatory response in contact dermatitis.41 The ‘nonceliac wheat sensitivity’ (NCWS) represents a further case for TNF family implication, in which the production of TNF-α by CD45+, CD3+, CD4+, and CD8+ cells, as well as of IL-17 by CD4+ cells in the rectal tissue of nonceliac wheat sensitivity patients significantly overpassed the controls.42 T helper (Th) 2 cells, together with Group 2 innate lymphoid cells (ILC2) drive allergic pathology under the effect of IL-13 and TNF-family cytokine TL1A that co-stimulates T cells through its receptor DR3.43 This receptor is required for ILC2 expansion and function in T cell-dependent and -independent models of allergic disease. In contrast, DR3-deficient ILC2 can still differentiate, expand, and produce IL-13 when stimulated by IL-25 or IL-33, mediating intestinal helminth expulsion.43
Therefore, some pyrogenic cytokines, especially TNF-α, contribute to various hypersensitivity disease development (Table 2). They comprise IgE-mediated, T cell-mediated, and some severe cutaneous DAs, also caused by NSAIDs. These pathologies involve a complex interaction mediated by diverse immune cells and mechanisms, affecting MCs, DCs, M2 macrophages, ILC2 cells, and specific T cell subpopulations.
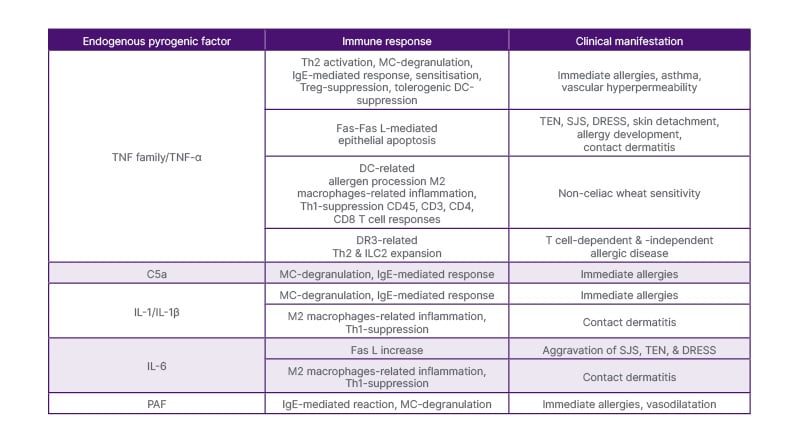
Table 2: Pyrogenic factors and allergic response.26-29,31,35,38-43
C5a: complement factor C5a; CD: differentiation cluster; DC: dendritic cell; DR3: T cell receptor DR3; DRESS: drug reaction with eosinophilia and systemic symptoms; Fas L: Fas ligand (Fas receptor); Fas: cell membrane molecule
Fas; ILC2: group 2 innate lymphoid cells; M2 macrophages: alternatively-activated macrophages; MC: mast cell;
PAF: platelet-activating factor; SJS: Stevens-Johnson syndrome; TEN: toxic epidermal necrolysis; Th: T helper cell.
Pyrogenic Factors, NSAIDs, Viral Infections, and Characteristics of Immuno-Allergic Response
When many people use a drug such as NSAIDs, adverse reactions can occur, conditioned by diverse genetic profiles, including the HLA, viral infections, or other underlying conditions.5,44 Responders can be selective or cross-intolerant, involving IgE or T cell immunologic mechanisms, COX-inhibitory pathway, or other (non)-immunologic mechanisms.7 Phenotypically, reactions can be classified as acute generalised exanthematic pustulosis, chronic spontaneous urticaria, contact dermatitis, DRESS, fixed drug eruption, NSAID-exacerbated cutaneous disease, NSAID-exacerbated respiratory disease, NSAID-induced urticaria/angioedema, single NSAID-induced delayed hypersensitivity reaction, single NSAID-induced urticaria, angioedema, or anaphylaxis, SJS, and TEN.
Besides promoting allergic responses,27,28,38,39,41 certain inflammatory mediators like IL-1β, IL-6, TNF-α, etc., may induce pyrogenic effects reaching amplified concentrations and more complex immune cell interactions (Figure 1).13-15,45 Thus, increased levels of TNF-α and IL-6 are observed in patients with COVID-19, especially in the severe group. Given the similarities of clinical features and pathogenesis between TEN and COVID-19, it is proposed that applying TNF-α inhibitor etanercept could attenuate disease progression in severe group COVID-19 patients by suppressing systemic auto-inflammatory responses.46 In the case of TEN, TNF antagonists are considered a controversial therapeutic alternative to large glucocorticoid doses or other immunosuppressors.33,35 The therapeutic use of anti-TNF-antibodies in autoimmune or viral pathologies is debatable because of T helper 2 (Th2) activation,26,30,47 and the amplification of similar immune responses in these cases may explain the induction of DA to NSAIDs during febrile infections.33,35 While the systemic use of NSAIDs or other drugs causes severe DA reactions like SJS or TEN, many etiological factors including herpes simplex virus are suspected in many cases of erythema multiforme minor and a solitary case of DRESS (caused by chikungunya virus).48-50
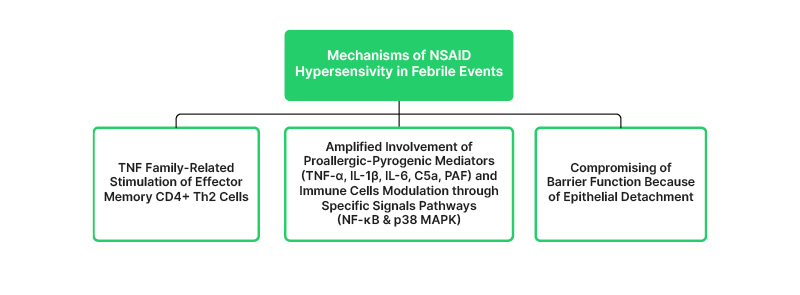
Figure 1: Mechanisms of non-steroidal anti-inflammatory drugs hypersensitivity in febrile events.14,15,26-31,33,38,39,41,42,45
CD: cluster of differentiation; C5a: complement factor C5a; NSAID: nonsteroid anti-inflammatory drugs; PAF: platelet-activating factor; Th:T helper cell.
The compromising of barrier function because of epithelial detachment in TEN and SJS represents an additional factor in the DA development after an infective event.33,35 In such circumstances, the organism may consider the intensified superficial contact with an allergen as exposure to a toxin.51,52 Like in other occasions, the immune response may switch via regulatory IL-10-dependent mechanisms.53,54 In SJS, epithelia-attached HIV leads to a dysregulated response by CD8+ T cells that recognise viral particles and their hyperactivation mimics the drug hypersensitivity reactions.55 Further viral reservoirs and first contact points in the mucosa are resident Langerhans and dermal dendritic cells. Consequent skin cytokine responses and dysregulated lymphoid populations create a crucible for hypersensitivity.56
Meanwhile, experiments in mice have shown that the TNF family costimulatory molecules OX40L/CD252 and CD30L/CD153 promote the reactivity of effector memory CD4+ Th2 cells after allergen exposure (Figure 1).45 In contrast, the blockade of costimulatory molecules induced a tolerogenic state and lack of eosinophilic inflammation. This indicates that several TNF costimulatory interactions may control the memory T cell responses and the severity of inflammatory reactions following (re)exposure to an allergen.45 Maybe, in this reported case, the coincident experience of viral fever and NSAID intake, among others, amplifies the TNF-related inflammatory mechanisms, including the ‘wake-up’ of memory CD4+ Th2 cells that lead to allergic symptoms. In contrast, the NSAID intake in non-febrile conditions does not amplify enough of the responsible mechanisms.
Apart from the crucial role in the immune protection against viral infections, the tissue-resident memory and regulatory T cells, once they cross-recognise the drug antigen, could be activated to attack surrounding epidermal cells through effectors, resulting in drug-induced tissue damage.57 A murine model of cytomegalovirus infection also manifested the antigen-virus interaction and virus-induced immune tolerance reduction, evidencing an allergic airway sensitisation only when airway infection has been combined with inhaled environmental antigens.58 Viral activation of airway mucosal CD11b+ DCs promoted their uptake and processing, thus, providing an ‘opened door’ for otherwise harmless ambiental agents. The airway re-exposure to the inhaled antigen induced a Th2 cell response, diverse mucosal pathohistological alterations, and amplified mucus secretion and airflow obstruction.58 Such dynamic data agree with the knowledge that, especially in children, most of the skin symptoms presumed as drug allergies are likely viral-induced or because of a drug-virus interaction. Usually, they do not personify a permanent, drug-specific, adaptive immune response (at least) to antibiotics.9,59
A study on mitochondrial oxidative stress shed additional light on the role of inflammatory cytokine IL-1β during NSAID exposure, an important PG-independent pathway that induces gastric mucosal injury. Indomethacin time-dependently stimulated the expression of proinflammatory molecules such as intercellular adhesion molecule 1, vascular cell adhesion molecule 1, IL-1β, monocyte chemotactic protein-1, and nuclear translocation of nuclear factor kappa-B (NF-κB) in gastric mucosa, in parallel with the increase of neutrophil infiltration and injury of gastric mucosa in rats.60 Meanwhile, the NF-κB signalling and the inhibition of regulatory TNF receptor 2 in CD4+ T cells aggravated the experimental airway inflammation in mice increasing the expression of cytokines IL-4, IL-5, IL-17, and TNF-α in serum and bronchoalveolar lavage fluid.61 Also, IL-3, IL-5, and GM-CSF could enhance p38 MAPK and NF-κB activity, and induce the expression of adhesion molecules intercellular adhesion molecule 1, CD11b, and CD18 on eosinophils during allergic inflammation.62,63 Inducing allergic exacerbation and eosinophilic inflammation, viral respiratory infections, and stimulation with poly (I: C) increase the percentage of CD11b+ cells and enhances the secretion of IL-8, with effects mediated via the p38 MAPK and NF-κB signalling pathways.64 In such situations, the additional increase of endogenous TGF-β expression impairs glucocorticoid anti-inflammatory action.63
In contrast to infection and NSAID effects, and despite the mentioned glucocorticoid insensitivity, its large doses suppress the NF-κB (and activator protein-1) signalling pathways, showing an anti-inflammatory effect. The interactions between NF-κB and the glucocorticoid receptor result in differing effects on histone acetylation and deacetylation.65,66 These data suggest that NSAIDs and viral infections activate inflammatory cytokines IL-1β, TNF-α, etc., which, via the p38 MAPK and NF-κB signalling pathways, could lead to allergic exacerbations and relative glucocorticoid insensitivity.
CONCLUSION
In conclusion, the collected data support the concept that, sometimes, NSAIDs may trigger ARs specifically in the presence of an inflammatory febrile response following viral infections, through a complex interplay of similar mechanisms. This mosaic may comprise the simultaneous involvement of proallergic-pyrogenic factors like C5a, IL-1β, IL-6, and especially TNF-α, in addition to the immune cells interaction that modulates the inflammatory response through the p38 MAPK and NF-κB signalling pathways. Additional ‘wake-up’ factors include the compromised barrier function because of epithelial detachment in TEN and SJS, and the possible implication of TNF family molecules that activate memory CD4+ Th2 cells after NSAID exposure during febrile conditions. In this case, an amplified TNF-related interaction may elicit the AR because of the simultaneous infective febrile response and consecutive NSAID intake. In contrast, NSAID administration for non-febrile conditions, or its avoidance during any febrile infection, does not recruit the immune system to mount the production of cytokines that provoke an acute allergic response.
Therefore, the literature findings (and the authors’ case report) indicate that regulatory and memory lymphocyte subpopulations can modulate the immune response toward allergic processes during inflammatory states (such as febrile viral infections). Maybe this condition, in combination with NSAID exposure, also provokes a (transient) decrease of the excitatory threshold for immune effector cells, thus inducing an AR. So, the infection and fever serve as cofactors for DA.
In this case study, the long COVID-19 and chronic tonsillitis could have contributed to this scenario. Considering the avoidance of responsible drugs as the best decision, the general scientific opinion accepts the use of aquatic baths at 29–33 °C and parenteral glucocorticoids as treatments of choice. Further alternatives are the use of COX-2 inhibitors and premedication with antihistamines or leukotriene modifiers that may help us in the controlled administration of an NSAID (as an add-on therapy to parenteral glucocorticoids). Concerning diagnosis, the drug challenge remains a gold standard test for the DA, but not all that glitter is gold. This case also teaches us that it is crucial to consider medical history thoroughly to ensure safe treatment. Future drug hypersensitivity classifications may incorporate such a fever-dependent ‘on/off’ (fed-on/off) mechanism (alias fever and drug-dependent ‘on/off’ allergic [feddonoffa] response). At the same time, such transient DA reactions need further investigation to understand comprehensively the underlying processes.






