
Abstract
Between 2006 and 2015, the U.S. Food and Drug Administration’s (FDA) overall likelihood of approval (LOA) from Phase I clinical trials for all therapeutic candidates was 9.6%, with the highest LOA in haematology (26.1%) and the lowest in oncology (5.1%). Two critical features attributed to the success of advancing trials were i) targeting driver genes responsible for disease, and ii) use of human disease-relevant animal models during preclinical studies. For decades, conventional wisdom has been that growth factors are the drivers of wound healing, but few have either advanced to clinical applications or proven effective. The purpose of this paper is to explore heat shock protein 90-alpha (Hsp90α)’s role as a potential driver of wound healing and as a possible future therapeutic entity through a review of recent literature, including studies with human disease-relevant animal models. Of the approximately 7,000 gene products generated by a given mammalian cell type, the Hsp90 family of proteins (Hsp90α and Hsp90β) accounts for 2–3% of them. Hsp90β fulfils the role of an intracellular chaperone, but Hsp90α’s intracellular function is surprisingly dispensable. Instead, the abundancy of Hsp90α appears to have been prepared for extracellular purposes. When secreted via exosomes by cells under environmental stress, such as injury, Hsp90α protects cells from hypoxia-induced cell death, reduces local inflammation, and subsequently promotes cell migration to repair the injured tissue. Unlike conventional growth factors, secreted Hsp90α stimulates all major cell types involved in wound healing equally, resists microenvironmental inhibitors like TGFβ and hyperglycaemia, and is highly stable. Inhibition of exosome-mediated Hsp90α secretion, neutralisation of Hsp90α’s ATPase-independent extracellular functions, or interruption of Hsp90α-LRP-1 signalling blocks wound closure in vivo. Topical application of Hsp90α’s therapeutic entity, F-5 (a 115-amino acid peptide), has shown great promise for healing acute burn and diabetic wounds in mice and pigs.
INTRODUCTION
According to the Wound Healing Society (WHS), approximately 15% of older adults in the USA suffer from chronic wounds, such as venous stasis ulcers, pressure ulcers (bedsores), and diabetic (neuropathic) foot ulcers, as well as other non-healing wounds caused by bacterial infections, ischaemia, and surgical procedures.1,2 Every year, 2–3 million Americans are diagnosed with various types of chronic wounds. Using diabetic foot ulcers (DFU) as a well-studied example, the impact of the disease burden of chronic wounds becomes staggeringly clear. Worldwide, 10–25 million diabetics develop DFU annually. In the USA, each of approximately 28 million diabetics has a 25% lifetime risk of developing a DFU.3-5 DFU are the leading cause of hospital admissions among diabetics and are associated with nearly 70% of all lower leg amputations, a morbidity that more commonly afflicts low-income individuals, members of minority groups, and those without health insurance.6 Unfortunately, the care currently available, which consists of antibiotics, routine debridement, negative pressure, hyperbaric oxygen, bioengineered skin equivalent grafting, and growth factor therapy, has shown either modest or little efficacy and at a very high cost. This paper explores the potential and reasoning behind the use of a previously unidentified therapeutic candidate, extracellular heat shock protein 90-alpha (Hsp90α), through a focussed review of recent literature.
THE WOUND CLOSURE PHASE OF WOUND HEALING
Wound healing is achieved through several overlapping processes: blood clotting (haemostasis), inflammation (immune system response), tissue growth (proliferation), and tissue remodelling (maturation). The first three processes complete within a few weeks after wounding and are collectively known as ‘the wound closure phase,’ the most critical and potentially life-threatening phase of wound healing. The remodelling phase following wound closure lasts at least a year and is difficult to follow in animal model experiments. Thus, the development of wound healing therapeutics has primarily targeted wound closure events; however, despite decades of effort, researchers have failed to identify the true driver genes responsible for promoting wound closure. The conventional target, a large serum polypeptide family known as growth factors, has led to minimal therapeutic innovation.7-9
Since the discovery of the first growth factor in the 1970s, locally released growth factors in injured tissues were believed to constitute the main driving force behind wound closure.10,11 Under this assumption, growth factors are responsible for inducing wound re-epithelialisation via promotion of the lateral migration of epidermal keratinocytes, as well as by subsequently acting as chemoattractants to induce the inward migration of dermal fibroblasts (which remodel damaged tissues) and microvascular endothelial cells (which revascularise the neodermis). The first growth factor, epidermal growth factor, entered clinical trials for treating partial thickness skin graft wounds in 1989.12 Subsequently, more than a dozen other growth factor trials were conducted, including i) epidermal growth factor treatment for traumatic corneal epithelial defects,13 chronically perforated tympanic membranes,14 and advanced DFU;15,16 ii) basic fibroblast growth factor (FGF) treatment for partial-thickness paediatric burn wounds,17 second-degree burns,18 and DFU;19 iii) acidic FGF treatment for partial thickness burns and skin graft donor sites;20 iv) granulocyte and macrophage colony-stimulating factor plus basic FGF treatment for pressure ulcers;21 and v) platelet-derived growth factor-BB (PDGF-BB) treatment for chronic pressure and diabetic ulcers.22-27 Most of these double-blinded trials reported promising clinical efficacy; however, only recombinant human PDGF-BB received the U.S. Food and Drug Administration’s (FDA) approval for treatment of DFU (becaplermin gel).28 Unfortunately, following its approval in 1997, multicentre, randomised, and parallel trials showed that becaplermin gel improved wound closure rates by 15% at best (50% improvement with treatment versus 36% with placebo).29-31 These outcomes were not considered cost-effective for clinical practice.23,24 Moreover, in 2008, the FDA added a black box warning to becaplermin gel regarding the increased risk of cancer mortality in patients requiring extensive treatments (≥3 tubes). PDGF-BB is one of the most potent natural mitogens identified and its dosage in becaplermin gel for clinical use is >1,000-fold the physiological PDGF-BB levels in human circulation.28 No other growth factors have since advanced to FDA approval.
WHAT WENT WRONG WITH THE CONVENTIONAL WISDOM?
To investigate this important question, the authors evaluated the biological activity of the FDA-approved PDGF-BB, becarplermin gel, as an example and found several possible explanations. Firstly, it is known that most growth factors engage distinct cell types selectively. Between keratinocytes, dermal fibroblasts, and microvascular endothelial cells (three cell types critical to successful wound healing), PDGF-BB can only engage PDGF receptor-positive dermal fibroblasts; neither keratinocytes nor endothelial cells express PDGF receptors.32 Secondly, members of the TGFβ family of cytokines present in the wound bed are known inhibitors of primary cell migration and proliferation and the authors have reported that TGFβ3 specifically nullifies the effectiveness of growth factors in the wound bed.33 Please note that this finding does not downplay the importance of TGFβ in extracellular matrix expression and deposition during the later phase of wound remodelling. Thirdly, pathological conditions, such as hypoxia (prevalent in wounds due to vascular disruption) and hyperglycaemia, further compromise the effectiveness of growth factors.32,34 In fact, vascular injury and obstruction in wounded tissue cause ischaemia, impairing growth factor delivery and creating a hypoxic microenvironment. These three hurdles, as illustrated in Figure 1, reduce the likelihood that growth factors are drivers of early-phase wound closure and lead the authors to speculate that the true driver lies elsewhere.35
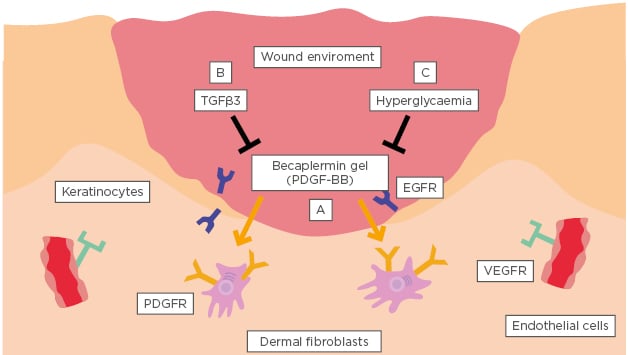
Figure 1: A conventional growth factor cannot be the driver for wound closure.
The three biological hurdles that prevent conventional growth factors from being effective during the early phase of wound closure are: A) selective targeting of only one cell type by any given growth factor; B) complete inhibition of growth factor functionality by concurrent presence of TGFβ in the wound bed; and C) brief, 2–10 minute half-lives within the unfavourable pathological microenvironment of the wound bed.
EGFR: epidermal growth factor receptor; PDGFR: platelet-derived growth factor receptor; PDGF-BB: platelet-derived growth factor subunit B (homodimer); VEGFR: vascular endothelial growth factor receptor.
DISCOVERY OF A NEW POTENTIAL DRIVER FOR WOUND CLOSURE: KERATINOCYTE-SECRETED HEAT SHOCK PROTEIN-90ALPHA
The authors postulated that the driver molecule probably originated from factors secreted by keratinocytes and/or fibroblasts under stress around the wound bed and, more importantly, that this factor must be able to overcome the three hurdles that make growth factors unlikely candidates. Using a sensitive cell motility assay as the standard of detection, protein purification from conditioned media of hypoxia-stressed primary human keratinocytes and dermal fibroblasts led to identification of the secreted form of Hsp90α.36,37 It was demonstrated that secreted Hsp90α i) acts as a pro-motility factor for all three skin cell types involved in wound closure, ii) is able to override TGFβ inhibition, and iii) remains fully functional even in the hypoxic and hyperglycaemic diabetic wound environment.38-40 Topical application of human recombinant Hsp90α protein accelerated full-thickness excision, burn, and diabetic skin wound closure by an average of 60% compared to the 15% reduction in closure time achieved by PDGF-BB (becaplermin gel, 0.01%) in mouse and pig models.32,34,38
A unique mouse model was generated in which the intracellular function of Hsp90α was selectively silenced by C-terminal deletion, but the extracellular function of Hsp90α, as well as constitutive expression of Hsp90β, was spared. These mice are phenotypically indistinguishable from wild-type (WT) counterparts.39 This mouse model allowed the authors to specifically assess the importance of Hsp90α’s extracellular non-chaperone function and determine whether it is essential for normal wound closure. The authors found that the chaperone-defective Hsp90α–∆ mutant mice experienced wound closure rates similar to Hsp90α-WT mice, suggesting that the secreted Hsp90α–∆ deletion mutant protein was fully functional in the promotion of wound closure. To prove this hypothesis, the authors cloned the mouse complementary DNA encoding Hsp90α–∆ and Hsp90α-WT and generated recombinant proteins. Topical application of Hsp90α–∆ mutant protein promoted wound closure as effectively as full-length Hsp90α-WT protein. Moreover, using 1G6-D7, an anti-Hsp90α monoclonal antibody developed in the authors’ laboratory,40 the authors showed that selective inhibition of extracellular Hsp90α–∆ protein function delayed wound closure in these mice.41 An earlier study by Song and Luo,42 who used a nude mouse model and neutralising antibodies against Hsp90, reported similar findings. Taken together, these studies provide direct evidence that extracellular Hsp90α acts as a potential driver for normal wound closure.
SECRETED HEAT SHOCK PROTEIN-90ALPHA IS NOT A CHAPERONE
Mechanistically, as illustrated in Figure 2A, secreted Hsp90α binds to subdomain II in the extracellular portion of the low-density lipoprotein receptor-related protein-1 (LRP-1) and activates the LRP-1 receptor. The NPVY motif in the cytoplasmic tail of LRP-1 connects extracellular Hsp90α signalling to serine-473, but not threonine-308, phosphorylation in Akt kinases. Akt1 and Akt2 work in concert to mediate extracellular Hsp90α’s cross-membrane signalling to promote cell survival, cell motility, and wound closure.43 The Hsp90α-WT protein, Hsp90α-E47D mutant, and Hsp90α-E47A and Hsp90α-D93N mutants have 100%, 50%, and undetectable ATPase activity, respectively.44
These constructs allowed Cheng et al.37 to test whether the chaperone-dependent ATPase is still required for the extracellular function of secreted Hsp90α. Using recombinant proteins encoded by these Hsp90α complementary DNA and cell motility assays, they reported that all the ATPase mutant proteins retained similar degrees of pro-motility activity compared to Hsp90α-WT. Moreover, these authors narrowed the functional entity of secreted Hsp90α to a 115-amino acid fragment, called F-5 (aa-236–aa-350), inside the linker region and middle domain. The extracellular F-5 fragment is visualised in three dimensions in Figure 2B. F-5 peptide alone promoted skin cell migration in vitro and wound closure in vivo as effectively as full-length Hsp90α-WT.32 These findings demonstrated that the N-terminal ATPase domain and the C-terminal dimer-forming and cofactor-binding domain are dispensable with regard to secreted Hsp90α’s ability to promote wound healing. Finally, Zou et al.40 compared amino acid substitutions between Hsp90α and Hsp90β (which has no extracellular functionality). Sequential site-directed mutagenesis allowed them to identify two evolutionarily conserved lysine residues, lys-270 and lys-277, in the Hsp90α subfamily that determine extracellular Hsp90α function. From zebrafish to humans, all Hsp90β subfamily members lack the dual lysine motif. However, substituting Hsp90β’s corresponding amino acid residues with lysines converted Hsp90β to behave like Hsp90α in vitro. These authors concluded that the N-terminal intrinsic ATPase and the C-terminal dimerisation region determine the intracellular chaperone function, whereas lys-270 and lys-277 determine the extracellular function of the Hsp90 protein family.
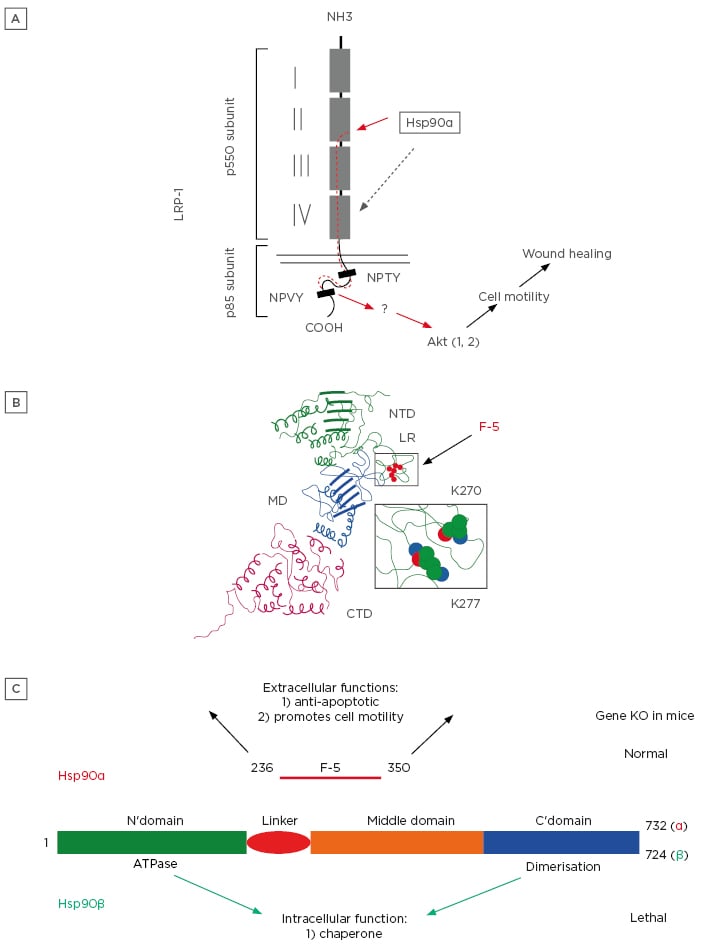
Figure 2: A fundamental and previously less recognised distinction between Hsp90a and Hsp90β.
A) Secreted Hsp90α binds to subdomain II in the extracellular portion of the LRP-1 and activates the LRP-1 receptor.
B) A three-dimensional visualisation of an extracellular F-5 fragment.
C) Under physiological conditions, Hsp90β is the critical intracellular chaperone for homeostasis, whereas Hsp90α is dispensable. Under pathological conditions, such as tissue injury, Hsp90α is secreted via exosomes to the extracellular environment. Once outside of cells, Hsp90α first prevents cells around the wound bed from undergoing hypoxia-induced apoptosis and, thereafter, promotes inward migration of keratinocytes, fibroblasts, and microvascular endothelial cells to close the wound.
Hsp90: heat shock protein-90; KO: knockout; LRP-1: low-density lipoprotein receptor-related protein-1.
REGULATION OF EXOSOME-MEDIATED SECRETION OF HEAT SHOCK PROTEIN-90ALPHA
Hsp90 proteins lack the signal peptide (SP) required for the classical endoplasmic reticulum–golgi protein secretory pathway. An alternative secretory pathway for molecules without SP is secretion through extracellular vehicles, such as exosomes. By comparing 43 intracellular signalling pathways for their potential roles in regulation of exosome secretion in human keratinocytes, Guo et al.45 identified the proline-rich Akt substrate of 40 kDa (PRAS40) as a pivotal signalling protein that connects environmental stress cues to exosome secretion machinery. This finding of PRAS40’s new role in exosome secretion is a significant departure from the understanding of PRAS40 established in the last decade.
Most previous studies focussed on PRAS40’s role in insulin signalling, as well that of other growth factors, in the mammalian target of rapamycin (mTOR) pathway (specifically mTORC1), which regulates cell metabolism, protein synthesis, and cell growth.46-51 These studies showed that, in growth-arrested cells, PRAS40 binds to mTORC1 via the raptor subunit and inhibits the kinase activity of the complex. Insulin stimulation activates Akt kinase via threonine-308 (Thr-308) phosphorylation. The Thr-308-phosphorylated Akt kinase in turn phosphorylates PRAS40’s C-terminus on Thr-246. Thr-246-phosphorylated PRAS40 dissociates from mTORC1, resulting in activation of mTORC1. The freed PRAS40 then (re-)associates with 14-3-3. However, PRAS40’s mechanism of action here does not explain the results of many other studies, which demonstrate that PRAS40 plays a positive role in tissue repair and tumourigenesis.52-56 The study by Guo et al.45 demonstrated that activated PRAS40 has little to do with the mTOR pathway, because the authors failed to detect any significant activation of mTORC1 in PRAS40-depleted cells. Instead, PRAS40 knockdown (KD) or PRAS40 dominant negative (DN) mutant overexpression blocks tissue injury signaling, including TGFα, hypoxia, and H2O2, and oncogene-induced exosome secretion in a variety of normal and tumour cells. Site-directed mutagenesis and gene rescue studies showed that Akt-mediated activation of PRAS40 via Thr-246 phosphorylation is both necessary and sufficient to cause exosome secretion, without affecting the endoplasmic reticulum–golgi pathway. Identification of PRAS40 as a linker protein paves the way for understanding how stress regulates exosome secretion under pathophysiological conditions, such as wound healing.
THE TWO MAJOR BIOLOGICAL FUNCTIONS OF SECRETED HEAT SHOCK PROTEIN-90ALPHA
Using purified recombinant Hsp90α proteins/peptides, two important biological functions of extracellular Hsp90α have been uncovered in both in vitro cell culture and in vivo animal models: protection of cell survival under hypoxic conditions and promotion of cell motility during wound closure. First, using a hypoxic model as an example of microenvironmental stress, cells were observed to secrete Hsp90α to prevent hypoxia-induced apoptosis.57,58 This finding is consistent with observed cellular behaviors in both physiological wound healing and cancer pathogenesis. During wound healing, cells in the hypoxic environment of the wound edge must remain viable prior to engaging in the repair process. Hypoxia and hypoxia-inducible factors (HIF)-1α are presently the best-characterised upstream stress cues that trigger Hsp90α secretion in both normal and tumour cells.36,59,60 The second important biological function of extracellular Hsp90α protein is its novel ability to act as a motogen without promoting mitogenic activity. Recombinant Hsp90α promotes migration, but not proliferation, of a wide range of cell types in the total absence of serum factors or any other exogenously supplemented large molecules.32,37
Tsen et al.43 demonstrated that the pro-survival and pro-motility functions of extracellular Hsp90α use a common signalling transduction pathway. According to their study, Hsp90α binds to subdomain II in the extracellular portion of the LRP-1. The NPVY, but not NPTY, motif in the cytoplasmic tail of LRP-1 connects Hsp90α signalling to Ser-473 (but not Thr-308) phosphorylation in Akt kinases, leading to enhanced cell migration. They further demonstrated that individual KD or knock-out of Akt1, Akt2, or Akt3 blocked extracellular Hsp90α signalling and wound healing in vitro and in vivo.43 Secreted Hsp90α’s promotion of cell survival and cell motility have been further verified in a distinct wound healing model. Bhatia et al.41 assessed the pro-survival effect of extracellular Hsp90α using a unique pathological burn wound process called ‘secondary burn wound progression’, in which the wound expands horizontally and vertically from the initial site of trauma. If left untreated, cells in the expanded areas soon die of necrosis, apoptosis, or both, due to ischaemia, infection, and accumulation of toxic metabolites. These authors showed that topical application of recombinant Hsp90α dramatically reduces the degree of secondary burn wound progression by preventing heat-induced cell apoptosis. Additionally, topically applied recombinant Hsp90α protein strongly stimulates wound closure by promoting keratinocyte migration-driven re-epithelialisation in mouse and pig wounds.32,34 Identification of these two functions of extracellular Hsp90α provides strong mechanistic support for currently ongoing therapeutic development of recombinant Hsp90α as a new treatment for various kinds of skin wounds.35 These two findings are depicted in Figure 2C. Based on these findings, and those of studies previously discussed, Hsp90α is not an essential intracellular chaperone and is only used under stress, such as by an injured tissue.
WHAT MAKES SECRETED HEAT SHOCK PROTEIN-90ALPHA A BETTER WOUND HEALING AGENT THAN GROWTH FACTORS?
The authors have identified three unique properties of extracellular Hsp90α that are absent in conventional growth factors. First, extracellular Hsp90α is a common pro-motility factor for all three types of human skin cells involved in wound healing because all three cell types express a compatible level of LRP-1, the receptor required for extracellular Hsp90α signalling.37 Following skin injury, keratinocytes migrate laterally to cover the open wound, and dermal fibroblasts and microvascular endothelial cells migrate inwardly to remodel the damaged tissue and re-establish a blood supply. Ideally, a single factor-based wound-healing agent should recruit all three types of skin cells into the wound bed. Secreted Hsp90α demonstrates this unique activity. Second, even in the presence of TGFβ3, extracellular Hsp90α remains equally effective in promoting migration of all three types of human skin cells. To the author’s knowledge, secreted Hsp90α is the first molecule identified that can override the inhibitory effects of the TGFβ family of cytokines. Third, all forms of diabetes are characterised by chronic hyperglycaemia, which is believed to be one of the major causes of delayed wound healing in diabetic patients. A reported mechanism of hyperglycaemia-induced wound healing impairment is destabilisation of HIF-1α protein, the key regulator of Hsp90α secretion.61,62 While hyperglycaemia blocks hypoxia-induced and serum-stimulated human dermal fibroblast migration, extracellular Hsp90α not only enhances hypoxia-driven migration in normal glycemic conditions, but also ‘rescues’ migration of cells cultured in hyperglycaemic conditions.32 In this case, extracellular Hsp90α may be promoting diabetic wound healing by bypassing HIF-1α down-regulation and jumpstarting migration of cells that could not otherwise respond to environmental hypoxia.
Keratinocyte migration occurs almost immediately following skin injury, whereas inward migration of dermal cells is not detected until 4 days afterward.7 During wound healing, cell migration precedes cell proliferation; when a cell is actively migrating, it cannot proliferate simultaneously. As cells at the wound edge move toward the wound bed, they leave ‘empty space’ between themselves and the cells behind them, which subsequently begin to proliferate after losing contact inhibition with the front of travelling cells.
The stimuli responsible for this subsequent stage of cell proliferation are likely plasma growth factors that have diffused from surrounding unwounded blood vessels, where TGFβ levels are low or undetectable. The role of secreted Hsp90α leads and expedites the initial wound closure event, enhancing motility to prevent infection, water loss, and severe environmental stress. This understanding is schematically depicted in Figure 3.
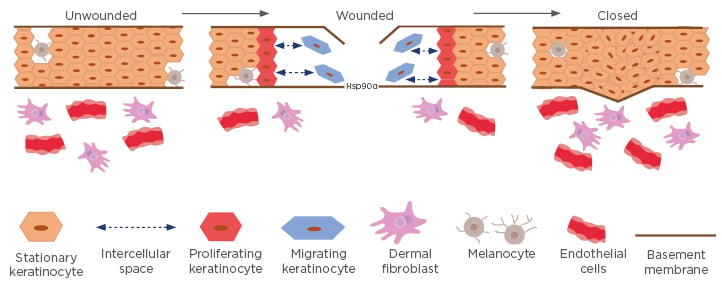
Figure 3: A model for migrating and proliferating keratinocytes around the wound edge.
In unwounded skin, cell–cell contact inhibition prevents both migration and proliferation. In wounded skin, keratinocytes (blue) at the wound edge secrete massive amounts of Hsp90α into the wound bed. The concentration of secreted Hsp90α reaches micromolar ranges within hours and serves as a chemoattractant for keratinocytes and dermal cells to migrate inward. The front of migrating keratinocytes creates space between themselves and the stationary keratinocytes behind them, resulting in the loss of contact inhibition that triggers the proliferation of the stationary keratinocytes to fill the gap. Together, these processes result in initial wound closure.
Hsp90: heat shock protein-90.
This concept, together with Figure 1, frames the argument that the driving force during early wound closure comes from keratinocyte-secreted Hsp90α, and not growth factors, which fail to overcome the three biological hurdles, are short lived, and are unable to reach the wound bed due to haemostasis.
IS EXTRACELLULAR HEAT SHOCK PROTEIN-90ALPHA A ‘GENERAL REPAIR MOLECULE’?
Recent studies have confirmed the previously contended notion that Hsp90α is not a critical intracellular chaperone like Hsp90β. Absence of Hsp90α or its intracellular chaperone form has a limited impact on mouse development,39,62 whereas Hsp90β knockout is lethal to developing embryos.63 At the cellular level, Zou et al.40 have recently shown that CRISPR/Cas9 knockout of Hsp90β led to cell death, whereas knockout of Hsp90α had little effect on cell viability. At the signaling level, Jayaprakash et al.57 reported that, under hypoxic conditions or conditions lacking serum factors, Hsp90β, but not Hsp90α, binds to the cytoplasmic tail of LRP-1 and stabilises the receptor at the cell surface. Hsp90α, but not Hsp90β, is then secreted by the same cells into the extracellular space, where it binds and signals through the LRP-1 receptor to promote cell motility, leading to wound closure. Regardless, the hypothesis that Hsp90α was designed by nature as a ‘general repair’ molecule is limited to indirect support and speculation, but the authors’ research has led them to view the large stockpiles of Hsp90α distributed throughout our bodies as something like fire stations strategically dispersed around a city; for the same reason that we build fire stations before fires occur, Hsp90α, unlike conventional growth factors, is pre-stored and locally released when nearby tissue is injured. In doing so, Hsp90α responds quickly and arrives equipped to extinguish TGFβ and hypoxia-induced inhibition. More work will be required to prove or disprove this hypothesis, but the authors look forward to exploring these new questions together, alongside the scientific community, with optimism and excitement.





