Abstract
Cancer treatment paradigms have evolved over recent years with an emphasis on personalised medicine. Targeted agents are being used to improve treatment outcomes and quality of life. For the treatment of non-small cell lung cancer, several agents with unique genetic and epigenetic targets are available. To this extent, mesenchymal–epithelial transition (MET), a heterodimer receptor tyrosine kinase involved in embryogenesis and organogenesis, has been investigated as a potential target for biological agents. MET dysregulation can occur via different mechanisms and trigger tumourigenesis and disease spread. Besides driving the oncogenic dependence of cells, MET is also involved in acquired resistance to epidermal growth factor receptor inhibitors. As such, many small molecule kinase inhibitors and antibodies have been developed or are currently in different phases of clinical trials to counteract the MET-induced neoplastic activity. Some of these agents are selective while others are nonselective with multiple other potential targets. This article aims to present an overview of biological functioning of MET, its role in oncogenesis and resistance to treatment, and clinical studies evaluating MET inhibitors for treatment of non-small cell lung cancer.
INTRODUCTION
There has been a decrease in the number of incidences of lung cancer recorded among men over the past few decades as well as among women in the last decade,1 which has been attributed to the advances in screening methods, surgical and radiation techniques, and new therapeutic modalities. In non-small cell lung cancer (NSCLC), many targetable receptors or protein kinases linked to complex cascades of signalling pathways have been identified to be oncogenic and/or dysregulated by oncogenic processes. The c-MET proto-oncogene, along with its protein product, MET (mesenchymal–epithelial transition; also known as hepatocyte growth factor [HGF] receptor) and cognate ligand is one such example of the potential drug targets that are being investigated in NSCLC and will be the focus of this review.
The MET proto-oncogene is located on chromosome 7 at locus 7q31. Its protein product is a 170 kD receptor tyrosine kinase, with highly glycosylated extracellular α-subunit and a transmembrane β-subunit, linked together by a disulfide bond (Figure 1).2
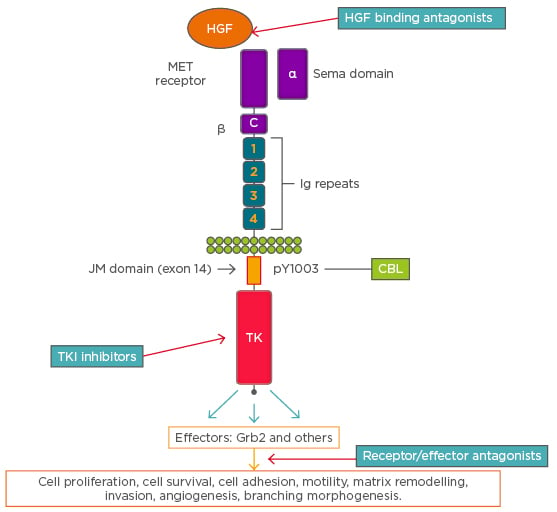
Figure 1: A schematic representation of cMET receptor and an overview of signalling and cellular effects. The figure also shows the sites of action of various cMET directed targeted agents along the receptor-signaling pathway.
C: cysteine-rich region; CBL: Casitas B-lineage lymphoma; HGF: hepatocyte growth factor; JM: juxtamembrane; MET: mesenchymal–epithelial transition; TK: tyrosine kinase domain; TKI: tyrosine kinase inhibitor. Adapted from Ma.2
The extracellular subunit contains the semaphorin domain, cysteine-rich MET-related sequence domain, and four immunoglobulin-plexin transcription domains. The β-chain contains a juxtamembrane domain, an intracellular tyrosine kinase domain that mediates MET biological activity, and a tail on the C-terminal, which is vital for the docking of substrates and downstream signalling.3 The juxtamembrane domain contains a serine residue (Ser 985), which inhibits the receptor kinase activity upon phosphorylation of a tyrosine molecule (Tyr 1003), which is responsible for MET polyubiquitination, endocytosis, and degradation upon interaction with the ubiquitin ligase Casitas B-lineage lymphoma (CBL).4 Two ligands of MET have been identified, mammalian HGF and scatter factor, along with their splicing isoforms and a bacterial leucine-rich surface protein named Internalin B.5 MET transcription is activated by HGF and other transcription factors, including hypoxia inducible factor-1 (HIF-1), leading to dimerisation and transphosphorylation of tyrosine residues in the intracellular domain that engages signal transducer proteins leading to downstream signalling. Some transducers, such as Grb2, Shc, Src, and the p85 regulatory subunit of phosphatidylinositol-3 kinase (PI3K), interact with the docking site of MET either directly or indirectly through the scaffolding protein Gab1.6,7 Phosphorylation of Gab-1 by MET interaction results in a sustained signal that mediates most of the downstream signalling pathways culminating in a range of biological activities, including mitogenesis, angiogenesis, morphogenesis, scattering, motility, migration, survival, and invasion.4,8 Different signal transduction pathways activated via MET engagement mediate different biological activities.
Ligand-independent activation of MET also occurs, which involves either the cell adhesion complex α5β1 integrin and fibronectin, or transactivation by other cell surface receptors, such as ErbB/HER family receptors or the interaction of Semaphorin 4D with its receptor Plexin B1.9,10
Cross talk between MET with other signalling pathways and the potential development of resistance to their receptor inhibitors is also well known. This is particularly seen in NSCLC where increased expression of epidermal growth factor receptor (EGFR)/HER family receptors directly leads to the transactivation of MET or vice-versa, and MET activation of Src indirectly leads to EGFR phosphorylation and creation of docking sites for EGFR transducers.9 MET has also been found to co-regulate oncogenic signalling pathways in collaboration with tumour suppressor genes (PTEN,11 VHL12) and promote angiogenesis and invasiveness of vascular endothelial growth factor (VEGF) via transcriptional upregulation of hypoxia-inducible factors.13
ROLE OF MET INHIBITION IN NON-SMALL CELL LUNG CANCER
MET Gene Mutations
A few gene mutations in the MET coding sequence have been found with varying degrees of relevance to oncogenesis. Somatic intronic mutation in the juxtamembrane domain, resulting in loss of CBL-E3 ligase binding, was characterised to demonstrate elevated MET expression and prolonged ligand-dependent MET activation.14 Normally, introns flanking MET exon 14 are spliced out, resulting in an mRNA transcript with an intact exon 14 coding for CBL-E3 ligase binding site. The mutation disrupts the splicing sites, resulting in aberrant splicing and skipping of exon 14 in the transcript. With the loss of the CBL-E3 ligase binding site, there is decreased polyubiquitination, delayed downregulation by the receptor, and sustained MET activation. Various exon 14 splice site alterations exhibiting diverse sequence composition that trigger constitutive activation of MET have been identified. More than half of these are indel mutations, many of which are novel. This diversity poses a diagnostic challenge and highlights the need for appropriate laboratory and analytical methods that can detect these alterations with high sensitivity and specificity. Approximately 3.5% of MET exon 14 alterations act as the driver mutation in lung adenocarcinomas, portending poor overall survival.15 MET exon 14 alterations are found in about 20.0% of pulmonary sarcomatoid carcinomas, a rare histologic subtype that is relatively refractory to conventional cytotoxic therapies.15,16 In a study of 933 patients with non-squamous NSCLC, patients with MET exon 14 alterations did not harbour activating mutations in KRAS, EGFR, or ERBB2, or rearrangements involving ALK, ROS1, or RET proto-oncogenes and as such were mutually exclusive to other oncogenic drivers.17 However, they can overlap with other alterations such as MET amplification or copy number gain and MDM2 proto-oncogene amplification.15 The incidence of this overlap is dependent on the definition of amplification used. The presence of MET exon 14 mutations was associated with advanced age and stage-dependent MET amplification, with a higher disease stage indicating a more likely presence of concurrent MET amplification. The presence of concurrent genomic alterations as potential modifiers of response to MET inhibition in MET exon 14–altered lung cancers will require exploration. Mutations in the MET tyrosine kinase domain have rarely been found in NSCLC as a secondary event resulting from exposure to prior therapies, such as tyrosine kinase inhibitors (TKI).18
In vitro, both small molecule TKI and MET-directed monoclonal antibodies have been found to be active in cell lines harbouring MET exon 14 alterations.14,17 These results have been replicated in the clinic, with a number of case reports of patients with lung adenocarcinoma harbouring exon 14 mutations with a clinical response to MET inhibition with TKI. In the majority of reports, the most frequent exon 14 alteration was splice donor mutation and partial responses to either selective or multi-targeted kinase inhibitors.14,16,17,19-21 This has further increased the impetus in drug development for these alterations and clinical trials investigating the therapeutic efficacy of both multi-targeted agents or selective MET kinase inhibitors are ongoing. No literature is yet available documenting clinical responses in these patients with the use of anti-MET or anti-HGF antibodies.
MET Gene Amplification
Amplification of the MET gene has been reported as the primary oncogenic event in 2–4% of TKI-naïve NSCLC and the secondary event in 5–20% in EGFR mutant NSCLC with acquired resistance to EGFR TKI.22 An increased copy number of the MET gene can be detected by fluorescence in situ hybridisation (FISH) or reverse transcriptase-PCR. MET gene amplification is expressed as the level of gene copy number gain (Capuzzo scoring system) and MET:CEP7 ratio. More than five signals per cell for copy number gain and a MET:CEP7 ratio >2 are both considered positive results for gene amplification. Setting the cut-off point for positivity too low may dilute the apparent drug benefit and fail to distinguish the true driver state.23 In the absence of oncogenic overlap with other drivers, lower ratios may signify a MET-associated phenotype. Nonetheless, MET amplification has been associated with a poor prognosis in patients with NSCLC24 and offers a targetable alteration.
Using MET gene amplification as a biomarker to predict response to MET inhibitors has been explored. In a Phase I expansion cohort, 13 MET-amplified NSCLC patients were treated with crizotinib.25 MET amplification status was determined by FISH and expressed as MET:CEP7 ratio. Patients were categorised into three groups based on the ratio: low with a MET:CEP7 ratio of ≥1.8–≤2.2 (n=1), intermediate for a ratio of >2.2–<5.0 (n=6), and high for a ratio of ≥5.0 (n=6). A response rate of 33% was reported (low n=0, intermediate n=1, and high n=3). It should be noted that MET exon 14 alterations harbour concurrent high-level MET copy number gain in approximately 20% of cases.15,17,19 In patients with EGFR mutations, secondary MET amplification leads to acquired EGFR TKI resistance by transactivating ErbB3 signalling.26 This has provided the rationale for various further clinical trials exploring the combination of MET and EGFR TKI in patients with mutant EGFR (Phase II expansion of TATTON trial,27 Phase II expansion of INSIGHT trial28).
MET Gene Fusion and Rearrangement
MET was first identified when the oncogenic chromosomal rearrangement Tpr-Met was induced in a sarcoma cell line.29 Though MET fusion gene products are not frequently found, they have recently been documented in lung adenocarcinoma. Stransky et al.30 demonstrated translocation events involving MET across different tumour types. Specifically in lung adenocarcinoma, fusion of the dimerisation motif to intact kinase domain led to generation of a chimeric fusion protein, KIF5B–MET. This novel fusion could explain part of the MET oncogenic activation and represent a potential therapeutic target.
Hepatocyte Growth Factor and Hepatocyte Growth Factor Receptor Overexpression and MET Activation
Studies have shown that higher levels of the HGF ligand can lead to aberrant MET activation and signalling independent of the changes at the receptor level.31 Increased expression of stromal-derived HGF was found to mediate VEGF-receptor (VEGFR) inhibitor-resistance and vascular remodelling in NSCLC by altering hypoxia-regulated molecules.32 Overexpression of the receptor itself without gene amplification, for example, by promoter demethylation, can result in MET upregulation.33 Similarly, modifications at the translational level can result in MET overexpression.34
Overexpression of HGF and activation of MET has been shown to confer resistance to chemotherapy agents. Specifically in NSCLC, in a study by Chen et al.,35 HGF was shown to induce cisplatin resistance via MET by activating focal adhesion kinase (FAK) and down regulating the expression of apoptosis-inducing factor. MET upregulation and increased secretion of HGF has been shown to occur in response to ionising radiation. In a preclinical study by Gao et al.,36 inhibition of MET led to radio sensitisation of cancer cell lines. Whether these processes occur in NSCLC post-radiation and mediate resistance and distant spread is currently unanswered.
MET INHIBITORS
Small Molecule Inhibitors
Capmatinib
Capmatinib is an ATP-competitive Type Ib selective inhibitor of MET that has shown anti-tumour activity by blocking c-MET dependent signalling and cross talk with EGFR and HER-3 in mouse xenograft models (Table 1).
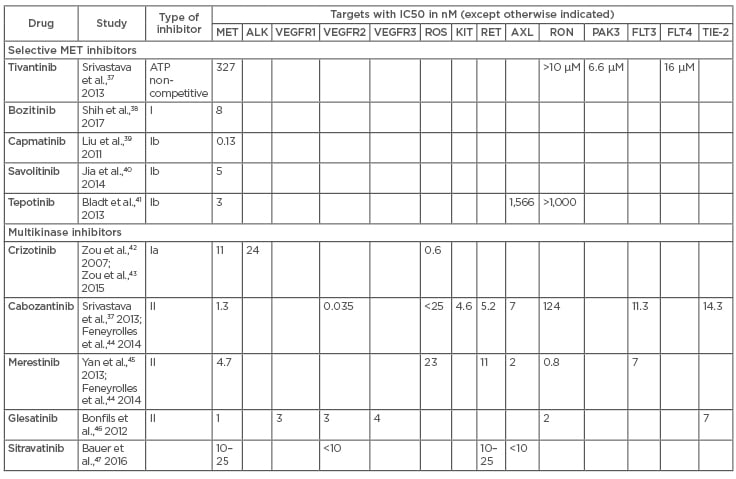
Table 1: Summary of IC50 data from available literature for oral MET TKI.
The table shows both selective and multikinase inhibitors along with the half maximal concentration of a particular inhibitor required for inhibition of its target molecule, expressed in nanomoles. MET: mesenchymal–epithelial transition; TKI: tyrosine kinase inhibitor.
In a Phase I study in patients with advanced solid tumours, including a cohort of EGFR-wild-type MET-mutated, amplified, or rearranged NSCLC patients, strong preliminary activity was observed in pre-treated patients with a high MET gene copy number (≥6) or MET overexpression of >3 as measured by immunohistochemistry (IHC). The objective response rate (ORR) was 47% and 24%, respectively. Significant tumour volume reduction of >45% was also observed in four patients with MET exon 14 alteration via molecular profiling.48 Capmatinib is being further investigated in a Phase II trial in NSCLC patients with MET exon 14 mutation (GEOMETRY mono-1)49 and in combination with erlotinib in patients with EGFR TKI-resistant NSCLC and acquired MET amplification (GEOMETRY duo-1).50
Tepotinib
Tepotinib is a Type Ib ATP-competitive selective MET inhibitor that has shown anti-tumour activity in patients with MET overexpressed or amplified NSCLC (Table 1). In a Phase Ib trial,51 combination of tepotinib and gefitinib was investigated in Asian patients with MET+/EGFR-mutant NSCLC. MET positivity was determined by IHC.51 Patients who had received a median of two prior lines of therapy, including an EGFR TKI, were enrolled (n=18). The combination was well tolerated with no dose-limiting toxicities reported and tepotinib 500 mg/day was confirmed as the recommended Phase II dose. Treatment-related adverse effects included increased blood sugar, amylase, and lipase levels, and neutropenia. Partial response was noted in 5 of the 18 patients (n=4 IHC 3+ and n=1 IHC 2+ tumours) and 4 of the 18 patients (IHC 2+, n=3) had stable disease. A Phase II trial of the drug is currently underway in patients with MET exon 14-altered NSCLC.52 Another Phase II trial of tepotinib plus gefitinib or cisplatin/pemetrexed in a 2:1 randomisation is also currently ongoing in patients with Thr790Met-negative and MET+ tumours who have failed first-line gefitinib.28
Savolitinib
Savolitinib is another Type Ib potent selective MET inhibitor that has shown anti-tumour activity in preclinical studies and entered Phase I and II studies (Table 1). In a Phase I study in patients with NSCLC, preliminary anti-tumour activity was observed in patients with increased MET gene copy number, gene amplification, or high MET protein expression.53 A Phase II trial of savolitinib is currently ongoing in patients with MET exon 14-positive pulmonary sarcomatoid carcinoma.54 It is also currently being investigated in a single-arm global Phase II study in combination with osimertinib based on encouraging data seen in the Phase I component.55
SAR125844 and Bozitinib (CBT101, PLB-1001, CBI-3103) are additional selective MET inhibitors in clinical development. Unlike the other kinase inhibitors discussed previously that are administered orally, SAR125844 is administered intravenously.56 It demonstrated modest activity, with low partial response rate of approximately 11% in patients with MET-amplified solid tumours. As a result, further development is currently halted. Bozitinib has demonstrated superior efficacy compared to crizotinib and capmatinib in preclinical lung cancer models.38 Several clinical trials are ongoing for bozitinib, including a Phase I study in patients with MET+ NSCLC.57
Crizotinib
Crizotinib is a multi-targeted kinase that was initially developed as a Type I MET inhibitor (Table 1) targeting resistance pathways to EGFR TKI; however, subsequent clinical development shifted upon the discovery of oncogenic ALK translocations in NSCLC, hence its first U.S. Food and Drug Administration (FDA) approval for this NSCLC genotype. Crizotinib was tested in patients harbouring MET-amplified metastatic NSCLC, with MET amplification by FISH used as the selection criteria for patients in a Phase I trial.25 Patients with intermediate and high levels of MET amplification showed significant tumour shrinkage with response rates of 17% and 67%, respectively. Further study of crizotinib in advanced MET-amplified NSCLC to optimise biomarker selection criteria is ongoing. More recently, the anti-tumour activity in patients with MET exon 14-altered NSCLC were enrolled into an expansion cohort of the Phase I PROFILE 1001 study. Partial responses were observed in 8 out of 18 patients, with an ORR of 44%, and 9 patients had stable disease.58 Enrolment of patients in this trial continues and further analyses are awaited. It is also being tested in the UK as part of the National Lung Matrix trial for patients with MET exon 14-altered lung cancer.59 Crizotinib in combination with erlotinib was also investigated as part of a Phase I trial.60 At a maximum tolerated dose of 150 mg of crizotinib, 1 of the 18 patients had partial response and 6 had stable disease. Reports of response to crizotinib in EGFR-mutant NSCLC patients are also emerging in the literature for those who have de novo MET mutations or acquire these during treatment with EGFR TKI, highlighting the role of MET alterations as mechanism for resistance to EGFR TKI and providing rationale for combination treatment. A significant clinical and radiological response was seen to the combination of erlotinib and crizotinib in a 70-year-old patient with EGFR-mutant NSCLC with de novo, high-level MET amplification who experienced primary resistance to erlotinib.61 Likewise, response to the combination of erlotinib and crizotinib was seen in a patient with primary EGFR-mutant and secondary MET-amplified NSCLC, though a novel crizotinib-resistant mutation MET Gly1108Cys eventually emerged.62
Cabozantinib
Cabozantinib is a multikinase Type II inhibitor of MET, ROS1, VEGFR, RET, KIT, and FLT3 (Table 1). It has been studied in a Phase II combination trial with erlotinib in patients with pretreated EGFR-mutant NSCLC patients who were found to have clinical activity, though MET amplification was not detected in any of the patients.63 Complete response to cabozantinib was reported in a case series of patients with concurrent MET exon 14-altered and MET-amplified NSCLC.19 In another case report, response to a combination of cabozantinib and erlotinib was reported in an EGFR and MET inhibitor pretreated patient who developed a new mutation in MET (Asp1228Val).64 Currently, this is being further investigated in a Phase II trial in NSCLC patients with RET fusion positive/ROS1/NTRK fusion/increased MET or AXL activity.65
Multiple additional multikinase MET inhibitors are undergoing Phase I/II studies in NSCLC patients. Glesatinib, a Type II inhibitor, has recently shown clinical activity in a NSCLC patient with MET exon 14 skip mutation upon development of resistance to crizotinib, with tumour response demonstrated in a Tyr1230His mutated metastasis.66 Sitravatinib, an inhibitor of MET, VEGFR2, PDGFRA, TAM family (AXL and MERTK), RET, NTRK1, and DDR2 has demonstrated favourable pharmacokinetic features, with plasma exposure exceeding the projected required levels for antitumour efficacy observed in preclinical studies.47 Merestinib targets ROS1, AXL, RON, MERTK, FLT3, DDR1/2, MST1R, and MKNK-1/2 in the nanomolar range, in addition to MET. In preclinical studies, it has shown several-fold more potent anti-proliferative activity against tumour cell lines with high MET gene amplification than the cell lines without MET amplification.45 Similarly, the study demonstrated a significant anti-tumour effect in two MET autocrine xenograft mouse models (U-87MG, KP4), a model with very high MET gene amplification (MKN45), and a model with MET overexpression and low MET amplification. Given its impressive multi-phenotypic anti-MET profile, it is being tested further in a Phase I study.67
Tivantinib
Tivantinib, thought to be a selective, non-ATP competitive inhibitor of MET, has been tested in many clinical trials (Table 1); however, subsequent studies have indicated that its efficacy is independent of MET signalling, for example through its ability to inhibit polymerisation of microtubules and induce cytotoxic effects.68-70 In a randomised Phase II trial evaluating activity of tivantinib in combination with erlotinib compared to erlotinib plus placebo in advanced NSCLC patients at progression, a trend was identified with regard to median progression-free survival (PFS) at 3.8 months in the experimental arm versus 2.3 months (p=0.24) in the placebo group.71 Pre-planned subgroup analyses revealed improvements for patients with non-squamous NSCLC, both in terms of PFS and overall survival (OS) with the combination treatment. Molecular exploratory analysis identified a trend with increasing magnitude of benefit that was directly correlated with level of MET gene copy number gain, although this did not reach statistical significance.
This provided the rationale for the subsequent Phase III MARQUEE study, comparing tivantinib plus erlotinib versus erlotinib alone in non-squamous NSCLC patients.72 However, the trial was terminated early due to negative results from a planned interim analysis of the primary end point of OS. During pre-planned subset analyses, PFS was higher with the combination treatment (3.6 months) compared to erlotinib alone (1.9 months; hazard ratio [HR]: 0.74; p<0.001) in the intention-to-treat population. Among patients with high MET expression tumours (as determined by a central laboratory using the Ventana SP44 rabbit monoclonal antibody), defined as membranous staining intensity of at least 2+ in at least 50% of tumour cells, a trend was identified that favoured OS in the combination treatment group (9.3 months) compared to the control group (5.9 months; p=0.03; HR: 0.70; 95% confidence interval: 0.49–1.01). Difference in PFS was similar to that observed for the whole intent-to-treat population. Tivantinib is currently being investigated in hepatocellular cancer but is not being further developed in NSCLC.
Anti-MET Receptor Antibodies
Onartuzumab
Onartuzumab is a humanised IgG1 monovalent anti-MET antibody with a single antigen-binding fragment (Fab) fused to a complete constant domain fragment (Fc) that inhibits high-affinity HGF binding to MET. Its efficacy was investigated in a Phase II study in combination with erlotinib versus erlotinib alone as second-line therapy for NSCLC patients.73 It was observed that 52% of patients who were MET positive by IHC analysis had a statistically significant reduction in the risk of disease progression by the addition of onartuzumab (median PFS: 1.5 versus 2.9 months; HR: 0.53; p=0.04). In the MET IHC-negative tumours, onartuzumab was detrimental. On further biomarker analysis, MET IHC was shown to be the most robust predictor of OS and PFS compared to other examined exploratory markers.74 Investigation of the drug in the Phase III Met Lung study failed to duplicate the positive results of the Phase II study and the trial was closed prematurely after an interim analysis showed no difference in OS.73 Additionally, patients with EGFR-mutant tumours in the experimental arm seemed to experience detrimental effects from the combination treatment on subgroup analysis. Onartuzumab is not currently being evaluated further as a treatment, though its use may potentially be repurposed through radiolabelling to assess the dynamic expression of MET in tumours as selection criteria for studies, such as clinical trials evaluating MET inhibitors in combination with EGFR TKI.75
Emibetuzumab
Emibetuzumab is a humanised anti-MET IgG4 monoclonal antibody that, despite its bivalency and thus potential to cause receptor dimerisation and activation, does not exhibit agonistic activity. It differs from onartuzumab because it also induces degradation of MET. It showed single agent clinical activity in a Phase I study of MET-amplified patients with advanced tumours, including NSCLC, with a disease control rate of 26% in patients with MET IHC >2.76 In another recent Phase I study, emibetuzumab in combination with erlotinib in pre-treated patients with advanced or metastatic NSCLC showed an ORR of 14.3% and a disease control rate of 28.6%.77 Emibetuzumab is currently undergoing Phase II studies in patients with NSCLC with EGFR mutations and in combination with ramucirumab in patients with advanced solid tumours including NSCLC.78 Several anti-MET antibodies, including LY3164530, JNJ-61186372, SAIT301, and ARGX-111, have shown preliminary activity in preclinical models and are being actively investigated in Phase I studies.79
Anti-Hepatocyte Growth Factor Antibodies
Ficlatuzumab
Ficlatuzumab is an anti-HGF antibody that binds to HGF and inhibits its binding to MET. In a Phase I trial of ficlatuzumab in patients with advanced solid tumours including NSCLC, stable disease was observed in 44% of patients who received the drug as monotherapy or in combination with erlotinib.80 In a Phase Ib study of ficlatuzumab combined with gefitinib in Asian patients with NSCLC, partial response and stable disease was noted in the 9 of the 15 trial participants.81 In the Phase II study, the combination of ficlatuzumab with gefitinib versus gefitinib alone was investigated in Asian patients with NSCLC. No statistically significant difference was observed regarding response rate or PFS between the two arms. However, on subgroup analysis, patients with EGFR mutations and low MET expression receiving the combination treatment showed an ORR of 41% versus 22% of the patients in single arm and median PFS of 11.0 months versus 5.5 months, respectively.82
In another Phase II study involving patients with NSCLC and EGFR activating mutations, the combination of ficlatuzumab and gefitinib did not improve clinical outcomes compared to the single agent gefitinib.83 Subgroup molecular analyses using VeriStrat, a proteomic platform, indicated that patients classified as ‘VeriStrat poor’ showed significant benefit from combination treatment in terms of PFS and OS, in both the intent-to-treat population and in the EGFR-mutant patients. However, a follow-up Phase II FOCAL study of ficlatuzumab in combination with erlotinib in NSCLC patients with a Veristrat-poor signature and EGFR mutations was terminated after interim analyses showed higher discontinuation rates than observed with the previous study.84
Rilotumumab
Rilotumumab is a fully human anti-HGF IgG2 antibody that has been investigated for the treatment of a variety of tumours due to its antineoplastic activity.85 Based on encouraging Phase II study results in patients with MET-positive gastric or gastroesophageal adenocarcinoma, two Phase III trials (RILOMET-1 and 2) were initiated. However, both trials were terminated after an interim safety review found rilotumumab had a lack of efficacy, including an increase in the number of deaths (128 deaths in rilotumumab arm versus 104 deaths in placebo arm) and significantly shorter median OS in the treatment arm (9.6 months versus 11.5 months; HR 1.37; p=0.016),86 irrespective of the level of MET expression. All subsequent trials, including a combination treatment arm with erlotinib for squamous cell NSCLC in the LungMap in NSCLC, have been terminated since late 2014.
RESISTANCE TO SMALL MOLECULE KINASE INHIBITORS
Several predominant mutations have been identified conferring resistance to small molecule kinase inhibitors in MET-dependent tumours. Acquisition or emergence of pre-existing clones with mutations in the MET activation loop Y1230 (also known as Y1248) or D1228 (also known as Y1246) have recently been shown to mediate resistance to type I kinase inhibitors such as crizotinib in NSCLC with MET exon 14 skip mutation.87 But sensitivity to type II kinase inhibitors such as cabozantinib is maintained,88 providing rationale for a clinical strategy of sequential therapy. Another cause of resistance is activation of the EGFR pathway due to increased expression of transforming growth factor α89 and inactivation of NF2 concomitantly with NRG1 overexpression.90
FUTURE DIRECTIONS
Researchers’ understanding of the biology and treatment of NSCLC with MET alterations continues to evolve. A multitude of agents utilising MET as a potential target have been tested or are under investigation at present. So far, the trials investigating these targeted agents have had mixed results, some promising and others negative. Over time, we have learnt that different MET alterations do not share the same clinical or functional equivalence and relevance. The selection of an appropriate patient population and optimal biomarker has been challenging, compounded by the myriad of methodologies and assays in the market that make analyses more complex. This has in part led to the variability in treatment outcomes among patients with MET alterations. To utilise the wealth of research that has gone into the understanding and development of MET inhibition and inhibitors, biomarker development should be validated more rigorously alongside efficacy evaluation. Rational drug combinations need to be developed to overcome resistance mechanisms. The pharmacological properties of novel agents have to be understood fully to gain insight into the toxicity profile and potential resistance mechanisms. Lastly, with the number of agents in development, a bottleneck has surfaced as a result of the limited number of biomarker-selected NSCLC patients who can qualify for the expanding number of trials testing these agents individually. Utilisation of multi-arm, multi-institutional umbrella trials permitting rapid inclusion of agents for clinical testing should be encouraged to accelerate and facilitate clinical evaluation and expand drug access for patients.






