
Abstract
Soft tissue sarcomas (STS) are a rare group of heterogeneous malignancies with >50 histologic subtypes that have varying biological behaviour and responsiveness to systemic therapy. The mainstay of therapy for metastatic STS in recent decades has been doxorubicin. To improve survival outcomes, numerous agents have been combined with doxorubicin; however, no combination has led to a survival benefit over doxorubicin alone until the recent use of olaratumab, a monoclonal antibody targeting platelet-derived growth factor-α. In addition to olaratumab, several other new drugs have surfaced as promising treatment options. Marine-derived chemotherapy agents, eribulin and trabectedin, are active in selecting STS subtypes. Both agents are effective in liposarcoma, while trabectedin also has activity in leiomyosarcoma. Further understanding of the importance of STS subtype-directed therapy, as well as the genomic complexities of STS, has led to development of several small molecule inhibitors for specific STS histologies. Agents targeting vascular endothelial growth factors, platelet-derived growth factors, and cyclin-dependent kinases 4 and 6 have all shown some efficacy in various STS subtypes. Similar to the selective activity of cytotoxic agents and small molecule inhibitors, immunotherapy, which has revolutionised management of numerous cancers, has also demonstrated activity in select STS subtypes. Collectively, these novel therapies highlight the importance of histology-directed approaches and of a greater understanding of the genomic landscape of STS. This review describes advances in chemotherapy, molecularly targeted, and immunotherapy agents for STS.
INTRODUCTION
Soft tissue sarcomas (STS) are a rare group of heterogeneous malignancies with >50 histologic subtypes that have varying biological behaviour and responsiveness to systemic therapy. Doxorubicin has been the mainstay of treatment in numerous subtypes of metastatic STS for decades, achieving response rates (RR) of 12–29% and average life expectancies of 12–18 months (Table 1).1-7 Until recently, numerous agents have been combined with doxorubicin with limited benefit. Olaratumab, a fully human IgG1 monoclonal antibody that targets platelet-derived growth factor receptor (PDGFR)-α, combined with doxorubicin resulted in a near doubling of overall survival (OS) compared with doxorubicin alone.4 Aside from doxorubicin-based regimens, novel chemotherapy agents, eribulin and trabectedin, have demonstrated efficacy in the L-sarcomas, liposarcoma (LPS), and leiomyosarcoma (LMS), highlighting the role of histology-directed therapy for these malignancies.
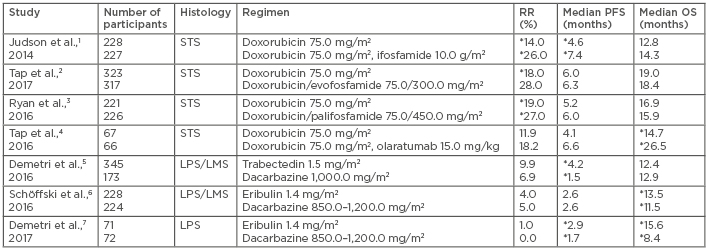
Table 1: Chemotherapy studies in advanced soft tissue sarcoma.
*Statistically significant.
LMS: leiomyosarcoma; LPS: liposarcoma; OS: overall survival; PFS: progression-free survival; RR: response rate; STS: soft tissue sarcoma.
Further understanding of the importance of subtype-directed therapy and the genomic complexities of STS has led to the development of small molecule inhibitors for certain STS histologies (Table 2).8-13 Tyrosine kinase inhibitors (TKI) (imatinib, sunitinib, and regorafenib) have dramatically changed the treatment landscape and outcomes for gastrointestinal stromal tumour (GIST), one of the most common STS subtypes.14-16 Imatinib, cediranib, and pexidartinib have also demonstrated activity in some of the rarest, chemo-refractory STS subtypes, including dermatofibrosarcoma protuberans,17,18 alveolar soft part sarcoma (ASPS),19 and tenosynovial giant cell tumour.20,21 The increased efficacy of these agents is due to the complex STS genomic landscape, including alterations to KIT, PDGFR-β, vascular endothelial growth factor receptors (VEGFR), and colony stimulating factor-1. Other more common STS also demonstrate potentially targetable aberrations in VEGFR, PDGFR-α and β, and cyclin-dependent kinases (CDK), suggesting a need for study of additional targeted therapies.
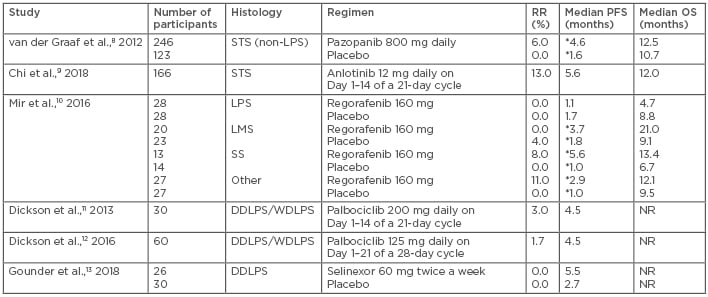
Table 2: Targeted agents in soft tissue sarcoma.
*Statistically significant.
DDLPS: dedifferentiated liposarcoma; LMS: leiomyosarcoma; LPS: liposarcoma; NR: not reported; OS: overall survival; PFS: progression-free survival; RR: response rate; SS: synovial sarcoma; STS: soft tissue sarcoma; WDLPS: well-differentiated liposarcoma.
Similar to the selective activity of cytotoxic agents and small molecule inhibitors, immunotherapy, which has revolutionised the management of numerous cancers, has also demonstrated activity in select STS subtypes. Immunotherapy trials have shown activity in undifferentiated pleomorphic sarcoma (UPS) and LPS (Table 3).22-25 Factors such as tumour microenvironment, tumour mutational burden, and the transcriptome have been associated with response to immunotherapy; however, the data for these factors in STS are limited.26,27 Correlative work to understand the relevant factors for predicting response in STS is ongoing. Numerous studies, including combinations of immunotherapy agents or immunotherapy combined with radiotherapy and/or chemotherapy, are also in progress.
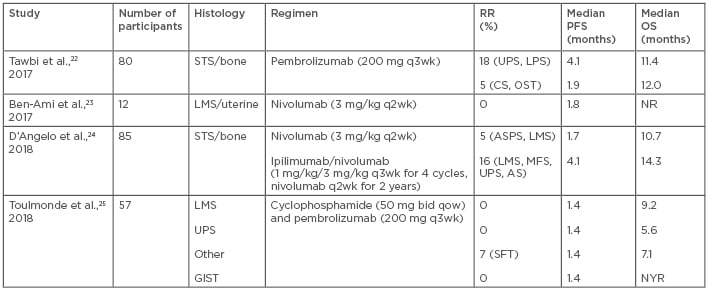
Table 3: Immunotherapy studies in sarcoma.
AS: angiosarcoma; ASPS: alveolar soft part sarcoma; bid: twice daily; CS: chondrosarcoma; GIST: gastrointestinal stromal tumour; LMS: leiomyosarcoma; LPS: liposarcoma; MFS: myxofibrosarcoma; NR: not reported; NYR: not yet reached; OS: overall survival; OST: osteosarcoma; PFS: progression-free survival; q: every; qow: every other week; RR: response rate; SFT: solitary fibrous tumour; STS: soft tissue sarcoma; UPS: undifferentiated pleomorphic sarcoma; wk: week.
As in other cancers, the understanding of the genomic complexity of STS has expanded over the past decade and spurred development of novel agents. The treatment paradigm for STS has shifted from treating all subtypes similarly towards a more histology-directed approach. In this review, the authors summarise recent developments in the treatment of non-GIST STS, as well as ongoing studies within the realms of chemotherapy, targeted therapies, and immunotherapy.
NOVEL CHEMOTHERAPY AGENTS AND COMBINATIONS
Doxorubicin has been the backbone of treatment for advanced STS for >40 years. The addition of ifosfamide (and its analogues evofosfamide and palifosfamide) and dacarbazine has resulted in improved RR but lacked a significant survival benefit and with increased toxicities.1,3,28-30 Olaratumab, a monoclonal antibody that targets PDGFR-α, was the first agent to be combined with doxorubicin and demonstrated an OS benefit in patients with metastatic STS. Olaratumab targets PDGFR-α by blocking the binding of PDGF ligands and preventing receptor activation. PDGFR-α is overexpressed in some STS subtypes31,32 and preclinical work in LMS cell lines has demonstrated antitumour efficacy,33 providing rationale for investigation in STS patients. A Phase I/II study4 enrolled varied STS subtype patients and randomised them 1:1 to doxorubicin with olaratumab versus doxorubicin alone. The progression-free survival (PFS) was 4.1 months and 6.6 months in the monotherapy and combination arms, respectively (p=0.0615), and the OS nearly doubled following combination therapy (26.5 months compared with 14.7 months with doxorubicin alone [p=0.0003]). The reason for the survival improvement with olaratumab remains unclear and preliminary analysis of the PDGFR-α expression status suggested no association with outcomes. Further investigation of the mechanism of action of olaratumab is needed to understand how the drug alters the tumour microenvironment and potentially improves the efficacy of doxorubicin. Olaratumab–doxorubicin combination therapy increased rates of neutropenia (58% versus 35%), mucositis (53% versus 35%), nausea (73% versus 52%), vomiting (45% versus 18%), and diarrhoea (34% versus 23%) compared with doxorubicin alone. However, despite increased neutropenia, there was no difference in the rates of febrile neutropenia or infection between the study arms. Infusion reactions, including two Grade 4 events, occurred in 13% of patients treated with combination therapy, but no cases were reported in those treated with doxorubicin alone.4 This study led to conditional approval of olaratumab in 2016 by the European Medicines Agency (EMA) and accelerated approval by the U.S. Food and Drug Administration (FDA) for the treatment of patients with STS not amenable to curative treatment with radiotherapy or surgery and with a histologic subtype for which an anthracycline-containing regimen is appropriate.
Data from the ANNOUNCE study,34 a Phase III trial comparing outcomes of STS patients treated with doxorubicin and olaratumab or doxorubicin alone, are expected in late 2019, and the results will determine whether the survival benefit withstands in a larger population. The results may also identify subtypes that have the greatest benefit and the mechanism of this survival benefit. Other ongoing studies are investigating neoadjuvant olaratumab as well as combinations with other sarcoma chemotherapy (gemcitabine–docetaxel,35 doxorubicin–ifosfamide,36 and doxorubicin–trabectedin37) and immunotherapy (pembrolizumab)38 agents, which may identify additional roles for olaratumab in STS. Trabectedin is a synthetically derived tetrahydroisoquinoline alkaloid originally isolated from the marine ascidian Ecteinascidia turbinata. Trabectedin binds to the minor groove of DNA, resulting in a conformational change of the DNA, bending towards the major groove and altering transcription regulation.39 The first Phase II studies of trabectedin in patients with advanced STS demonstrated a RR of 4–17%, median PFS of 1.9 months, and median OS of 9.2–12.8 months.40-43 Given the paucity of treatment options for STS and the clinical activity and tolerability, the drug received approval by the EMA in 2007 for the treatment of patients with advanced STS after failure of anthracyclines and ifosfamide or for those who were unfit to receive these agents.
In these initial Phase II studies, patients with L-sarcomas, particularly LMS and myxoid round cell LPS, showed the greatest benefit.40-44 Therefore, a multicentre Phase III trial45 compared trabectedin 1.5 mg/m2 every 3 weeks to dacarbazine 1,000 mg/m2 every 3 weeks in L-sarcoma patients after prior anthracycline treatment and at least one additional regimen. The median PFS was improved, measuring 4.2 months versus 1.5 months (p<0.001) with trabectedin and dacarbazine, respectively. In addition, the median PFS improvement was greatest in the myxoid round cell LPS group, totalling 5.6 months versus 1.5 months with trabectedin and dacarbazine, respectively. There was no difference in RR or OS. Trabectedin was also well tolerated, with the most common serious adverse events (AE) being myelosuppression and transient liver function test elevation.5 This study led to FDA approval for trabectedin in 2015 for the treatment of patients with unresectable or metastatic LPS or LMS who had received a prior anthracycline-containing regimen. Trabectedin became the third FDA-approved drug for STS treatment after doxorubicin (1974) and pazopanib (2012).45 The approval of trabectedin, based on improvement in PFS, demonstrates the acceptance of disease stability as a meaningful endpoint in metastatic STS and highlights that disease response by Response Evaluation Criteria in Solid Tumors (RECIST) is uncommon in STS, underscoring the need for novel systemic therapies.
Eribulin mesylate is a synthetically derived analogue of halichondrin B, which was originally derived from a marine sponge. It is a non-taxane microtubule inhibitor that prevents mitotic spindle formation, inducing cell cycle arrest.46 Eribulin was initially FDA-approved to treat advanced or metastatic breast cancer. Similar to trabectedin, a Phase II study47 of eribulin in multiple STS subtypes demonstrated activity solely in the L-sarcomas. The proportion of LMS and LPS patients who were progression-free at 12 weeks was 31.6% and 46.9%, respectively, which compared favourably to historical controls.47 Due to the activity in L-sarcomas, a Phase III study6 compared the efficacy of eribulin 1.4 mg/m2 on Days 1 and 8 of a 21-day cycle with dacarbazine 850–1,200 mg/m2 every 3 weeks only in these patients. The OS was improved, totalling 13.5 months versus 11.5 months with eribulin and dacarbazine, respectively (p=0.0169), and there was no difference in PFS or RR.6 In a preplanned subgroup analysis, the primary benefit of eribulin was in LPS, with improved OS (15.6 months versus 8.4 months, respectively [p<0.001]) and PFS (2.7 months versus 1.9 months, respectively [p=0.0015]) in the eribulin group compared to the dacarbazine group. Notably, there was no difference in RR.7 In both cohorts, eribulin was associated with a greater incidence of AE Grade ≥3 (67%) than dacarbazine (56%). Most severe AE were haematologic; however, the incidence of neutropenic fever was low.6 Collectively, these data led to the approval of eribulin by the EMA and FDA in 2016 for patients with unresectable or metastatic LPS after a prior anthracycline-based regimen, but not for patients with LMS. This agent provides a reasonable second-line option for treating advanced LPS because it demonstrated a 2-month survival benefit and was reasonably well tolerated.
TARGETED AGENTS
Over the past two decades, molecularly targeted agents have emerged as effective anti-cancer therapies. The success of imatinib and trastuzumab in revolutionising the treatment of chronic myelogenous leukaemia and HER-2-positive breast cancer, respectively, sparked greater analysis of cancer genomics and evaluations of how genetic abnormalities could be used in developing novel anticancer strategies. STS demonstrate overexpression and/or mutations of numerous potential therapeutic targets. For example, overexpression of VEGF has been associated with higher-grade tumours and worse outcomes in sarcoma patients, and targeting VEGF has been explored as a potential therapeutic strategy with reasonable efficacy.48-50 Increased expression of CDK4, CDK6, and PDGF, as well as mutations in PDGFR-α and β, have also been described in STS. These molecular abnormalities have provided justification for several studies, which are described in greater detail later in this review.4,8,32,51
Pazopanib is an oral, synthetically derived indazole pyrimidine that inhibits VEGFR 1–3, PDGFR-α and β, and c-kit.52 VEGF and PDGF are factors in STS angiogenesis, providing a rationale to study pazopanib as a treatment option. An initial Phase II study evaluated daily 800 mg pazopanib in 142 advanced STS patients in a Simon two-stage design.53 Patients were stratified into four cohorts: adipocytic sarcoma, LMS, synovial sarcoma (SS), and other sarcomas. The adipocytic cohort closed after the first stage, but the other three cohorts completely accrued. The PFR at 12 weeks for the LMS, SS, and other sarcomas was 44%, 49%, and 39%, respectively. PFS and OS compared favourably to historical controls; in the LMS, SS, and other sarcomas, the median PFS and OS were 91 and 354, 161 and 310, and 91 and 299 days, respectively.53 These data provided the basis for the Phase III study of pazopanib in patients with advanced STS except the adipocytic subtypes.8 Patients were randomised 2:1 to receive either daily 800 mg pazopanib or a placebo. The median PFS was improved with pazopanib, recorded as 4.6 months versus 1.6 months with placebo (p<0.0001). However, the OS was not significantly different with pazopanib and placebo (12.5 months versus 10.7 months, respectively [p=0.25]) and the RR were 6% with pazopanib and 0% in the placebo group. The most common severe AE were fatigue (13%), hypertension (7%), anorexia (6%), and diarrhoea (5%).54 Overall quality of life was not significantly worsened by pazopanib.54 This study led to the approval of pazopanib in 2012 by the EMA and the FDA for patients with advanced STS, except adipocytic sarcomas, who have received previous chemotherapy. This approval again demonstrates the value of stable disease in the treatment of metastatic STS.
Anlotinib is an oral multikinase inhibitor that targets VEGFR 2 and 3, fibroblastic growth factor receptor 1–4, PDGFR-α and β, c-kit, Ret, Aurora-B, c-FMS, and discoidin domain receptor 1.55 An initial Phase II study of this agent included 166 sarcoma patients who received a daily 12 mg anlotinib dose in a 2-week-on and 1-week-off regimen.5 The ORR was 13%. Responses were seen in 8% (2/26) of LMS, 11% (2/18) of fibrosarcoma (FS), 17% (8/47) of SS, 46% (6/13) of ASPS, and 14% (1/7) of clear cell sarcoma patients. The overall median PFS and OS were 5.6 months and 12.0 months, respectively.9 A Phase III study of this agent randomised 233 patients with SS, LMS, and ASPS to either anlotinib (n=158) or placebo (n=75). The median PFS was 6.3 months for anlotinib versus 1.5 months for placebo (hazard ratio: 0.33; p<0.0001). The PFS improvement was greatest in the ASPS cohort, recorded as 18.2 months versus 3.0 months (hazard ratio: 0.14; p<0.0001) with anlotinib and placebo, respectively. The ORR was 10.1% for anlotinib versus 1.3% for placebo (p=0.0145). The most common Grade ≥3 AE were hypertension (19%), gamma glutamyl transferase elevation (4.4%), triglyceride elevation (4.4%), low-density lipoprotein elevation (3.2%), hyponatraemia (3.2%), and neutrophil count reduction (3.2%).56 Overall, anlotinib is well tolerated and its use is promising in multiple STS subtypes. It is currently being evaluated in a Phase III study versus dacarbazine in LMS, SS, and ASPS patients.57
Regorafenib is an oral TKI that targets VEGFR 1–3, PDGFR, KIT, RET, and Raf, and is EMA and FDA-approved to treat GIST, colorectal cancer, and hepatocellular cancer. REGOSARC10 was a double-blind, placebo-controlled Phase II study of four cohorts of STS: LPS, LMS, SS, and other sarcomas. Patients were randomly assigned to receive 160 mg daily regorafenib on Days 1–21 of a 28-day cycle or placebo. There were no significant differences in RR or OS; however, the median PFS was significantly improved in all cohorts except for LPS. The most common Grade 3/4 AE included asthenia (13%), hand and foot skin reaction (15%), hypertension (19%), and hypophosphataemia (13%). There was one Grade 5 hepatitis-induced liver failure that was related to regorafenib.10 This study demonstrates the activity of regorafenib in non-adipocytic STS, and further investigation in a Phase III study against an active agent is warranted.
The TKI described thus far have limited or no activity in adipocytic sarcomas, suggesting that alternative targets are needed. Palbociclib is an oral inhibitor of CDK4 and CDK6 that prevents phosphorylation of the retinoblastoma protein and can result in tumour stasis or regression.58 CDK4 is overexpressed in two subtypes of adipocytic sarcoma, well-differentiated LPS (WDLPS) and dedifferentiated LPS (DDLPS), as compared to normal fat cells.59 Preclinical work demonstrated the antitumour activity of palbociclib in WDLPS/DDLPS cell lines and in xenografts.60 Two Phase II studies11,12 confirmed antitumour activity of palbociclib in 90 patients with WDLPS/DDLPS. The studies evaluated different dosing regimens, either 200 mg daily on Days 1–14 of a 21-day cycle (high dose) or 125 mg daily on Days 1–21 of a 28-day cycle (low dose). The primary endpoint was met in both studies, achieving a PFS at 12 weeks of 66% in the high-dose and 57% in the low-dose group. The median PFS was 18 weeks in both studies and the low dose was slightly better tolerated. Grade 3/4 AE were primarily haematologic: anaemia (17% versus 22%), thrombocytopenia (30% versus 7%), neutropenia (50% versus 36%), and febrile neutropenia (3% versus 0%) with the high and low doses, respectively.11,12 Correlative work from paired tumour biopsies demonstrated that benefit from palbociclib treatment was associated with downregulation of MDM2,61 suggesting a potential biomarker that could be used to predict response to CDK4 inhibition.
Another potential novel treatment approach for DDLPS is selinexor, an oral selective inhibitor of nuclear export that binds to the nuclear export protein XPO1. This causes tumour suppressor proteins to accumulate in the nucleus, resulting in selective destruction of cancer cells while sparing the healthy cells. A Phase II study evaluated selinexor 60 mg twice a week in 56 patients with advanced DDLPS. The primary endpoint was PFS and selinexor demonstrated a trend towards improved PFS over placebo (5.5 months versus 2.7 months; p=0.26). Treatment was well tolerated, with the most common Grade 3/4 AE being hyponatraemia (19.2%), anaemia (19.2%), thrombocytopenia (11.5%), neutropenia (7.7%), and hyperglycaemia (7.7%).13 The Phase III portion of the study is still ongoing and is comparing selinexor to placebo in patients with advanced DDLPS.62
IMMUNOTHERAPY
Immunotherapy was first described as a potential anticancer strategy in the 19th century in sarcoma patients. Streptococcal antigens (Coley’s toxins) were injected into sarcomas and resulted in tumour shrinkage.63 However, there was doubt about these findings, and investigation of chemotherapy and radiation took precedence over further investigation of immunotherapeutic options. More recently, immunotherapy agents targeting T cell checkpoint molecules, such as cytotoxic T lymphocyte-associated protein 4 and programmed death receptor (PD-1) and its ligand (PD-L1), have revolutionised the treatment of numerous malignancies.64-68 However, the success of immunotherapy agents in sarcoma in the modern era has been limited (Table 3).
One of the initial investigations of immunotherapy in sarcoma was SARC028,22 a two-cohort, single-arm, open-label Phase II study of pembrolizumab, an anti-PD-1 monoclonal antibody, administered intravenously (IV) every 3 weeks at a dose of 200 mg. Forty patients were enrolled into each of the bone and soft tissue cohorts. The STS cohort was split into 10 patients with each of the following histologies: UPS, LPS, LMS, or SS. RR were highest in the UPS and LPS cohorts, measuring 40% and 20%, respectively. No responses were seen in LMS patients. In the bone cohort, the RR were 5% (1 out of 22) in osteosarcoma, 20% (1 out of 5) in chondrosarcoma, and 0% (0 out of 13) in Ewing’s sarcoma. The median PFS and OS were 18 and 49 weeks in the STS cohort, and 8 and 52 weeks in the bone cohort, respectively. Treatment was well tolerated, with treatment-related serious AE occurring in 11% of patients. AE included pneumonitis (4%), adrenal insufficiency (4%), pulmonary embolism (2%), interstitial nephritis (2%), infectious pneumonia (2%), bone pain (2%), hypoxia (2%), and pleural effusion (2%). There were no Grade 5 AE. The study concluded that pembrolizumab was promising in UPS and LPS and recently completed enrolment of additional patients into these cohorts. Select results from correlative work were included in the initial analysis. PD-L1 expression was identified in 5% (2 out of 40) of the STS samples, both cases were from UPS patients who had responded to therapy. However, responses were also noted in non-PD-L1-expressing LPS patients, suggesting that predicting response to anti-PD-1 therapy is based on more than PD-L1 expression.22 Additional correlative work from this study is pending and will offer further insight into the role of immunotherapy in sarcoma.
A smaller study23 evaluated nivolumab, an anti-PD-1 monoclonal antibody, 3 mg/kg IV every 2 weeks in patients with metastatic uterine LMS. Twelve patients were enrolled and no responses were seen, suggesting a lack of benefit and precluding further enrolment. The median PFS was 1.8 months and a median OS was not reached. Treatment-related serious AE occurred in 25% of patients, with solitary cases of abdominal pain, elevated amylase and lipase, and fatigue. Correlative work demonstrated PD-1 and PD-L1 expression in 20% of samples, but no correlation with outcomes was observed.23 In combination with the findings from SARC028, this study further demonstrates the lack of efficacy of anti-PD-1 monotherapy in LMS. LMS resistance may be due to the density of tumour-associated macrophages, PTEN mutations, and reduced expression of genes encoding neoantigens.69,70 However, recent translational work suggests that LMS is an inflamed tumour type with high levels of T cell-related gene expression and occasional strong expression of PD-L1, indicating that immunotherapy may be effective but that a combination strategy may be a better approach.71
Combining immunotherapy agents, such as ipilimumab, an anti-cytotoxic T lymphocyte-associated protein 4 monoclonal antibody, and nivolumab, is an effective strategy in melanoma and renal cell carcinoma.72,73 As a result of the potential synergy of these agents, a Phase II study evaluated two treatment strategies: nivolumab with or without ipilimumab in sarcoma. Treatment included 3 mg/kg IV nivolumab every 2 weeks or 1 mg/kg IV ipilimumab with 3 mg/kg nivolumab every 3 weeks for four doses, followed by 3 mg/kg nivolumab every 2 weeks for up to 2 years. The study was not designed to compare results between treatment arms. The RR was 5% with nivolumab and 16% with combination therapy. In the monotherapy arm, responses were seen in ASPS and non-uterine LMS, while, in the combination treatment arm, responses were seen in LMS (n=2), UPS (n=2), myxofibrosarcoma, and angiosarcoma. The median PFS and OS were 1.7 and 10.7 months and 4.1 and 14.3 months with monotherapy and combination therapy, respectively. Given that the monotherapy did not reach its target RR, nivolumab alone is considered inactive; however, the combination has activity similar to other approved sarcoma therapies and is being further investigated in UPS and LPS. Treatment-related serious AE occurred more frequently with combination therapy (26% versus 19%) than in the monotherapy arm. AE included adrenal insufficiency, elevated alanine and aspartate aminotransferase, hyponatraemia, anaemia, fatigue, pain, and pruritus with dual agent therapy, and anaemia, thrombocytopenia, anorexia, dehydration, diarrhoea, fever, elevated creatinine, and pleural effusion in the monotherapy cohort. Correlative work, including PD-L1 expression, tumour-infiltrating lymphocytes, mutational burden, neoantigen analysis, and T cell receptor clonality, is in progress.24 Results of these studies will help determine factors that predict response or suggest a role for further study of combination immunotherapy in sarcoma.
Adding chemotherapy, targeted therapy, or radiation therapy to immunotherapy to augment efficacy is an area of active investigation. Combining axitinib, a pan-VEGFR inhibitor, with pembrolizumab has showed promise in treating ASPS. The 3-month PFS rate was 90.9% (95% confidence interval: 50.8–98.7) and ORR was 45.5% (95% confidence interval: 18.1–75.4). Correlative studies found high plasma angiogenic activity, a circulating neutrophil:lymphocyte ratio <4.1, low naïve fraction CD4+ tumour-infiltrating lymphocytes, and low PD1+CD8+ peripheral blood mononuclear cell were associated with lack of progression. Overall, this combination was well tolerated and demonstrated activity in ASPS, warranting further study.74
Given the potential immunomodulatory effects of metronomic cyclophosphamide and its activity in STS treatment, the French Sarcoma Group combined oral cyclophosphamide 50 mg twice daily every other week with 200 mg pembrolizumab IV every 3 weeks in four cohorts: LMS (n=15), UPS (n=16), GIST (n=10), and other sarcomas (n=16).25 There was one partial response in a patient with solitary fibrous tumour and the median PFS was equal across cohorts at 1.4 months. The OS varied and was 9.2 months, 5.6 months, 7.1 months, and not yet reached in the LMS, UPS, other, and GIST cohorts, respectively. Correlative work demonstrated PD-L1 expression in immune cells was 23%, 64%, 29%, and 43% in the LMS, UPS, other, and GIST cases, respectively.25 The only patient with immune cell PD-L1 expression >10% was also the only patient who responded to therapy. Additional translational studies evaluated expression of CD8, CD68, CD163, and IDO1. However, given the lack of reference values for these markers in sarcoma, the findings were difficult to interpret. Results were compared to a dataset derived from non-small cell lung cancer patients and revealed that CD8 densities were significantly lower in sarcoma patients compared to non-small cell lung cancer patients. Also, high infiltration by CD163+ macrophages and by macrophages that expressed IDO1 was seen in sarcomas, which potentially provides a mechanism for the PD-1 resistance seen in these tumours. An increased plasma kynurenine:tryptophan ratio correlated with increased IDO1 expression, adding further support to the IDO1 pathways as a mechanism of resistance to anti-PD-1 therapy.25
CONCLUSION
STS are a highly heterogeneous group of tumours with varying responses to treatment. Given their variable genomic makeup, histology-directed therapy should be regarded as the future of treatment. Currently, combined doxorubicin and olaratumab is the first-line treatment regimen for numerous STS subtypes; however, results of the Phase III study may discern subtypes that derive the greatest benefit. Trabectedin and eribulin have demonstrated efficacy in the L-sarcomas, but further investigation is needed to understand why these subtypes have the greatest success. Targeted therapies, such as pazopanib, have an established role in treating non-adipocytic STS. Novel agents, anlotinib, palbociclib, and selinexor, have shown promise in Phase II studies; however, larger, confirmatory Phase III studies are awaited to determine whether new options for LMS, SS, ASPS, and DDLPS will become available. The role of immunotherapy in STS remains uncertain and is currently only recommended within the context of a clinical trial. Responses in UPS and LPS are encouraging; however, additional studies evaluating more patients, combination strategies, and correlative work are needed. Collectively, the results of recent studies demonstrate the ability of the sarcoma community to enrol histology-tailored trials, which will allow for the development of more subtype-specific therapies.






