
Abstract
Leprosy is a model disease for understanding human immune responses underlying diseases caused by intracellular pathogens, as well as providing valuable insights into autoimmune disorders and cancer. This review addresses the unresponsiveness/anergy of host T cells to the causative pathogen Mycobacterium leprae and describes both the adaptive and innate immune responses observed during the clinical course of the disease. Leprosy presents as a clinicopathological spectrum, with divergence in antigen-specific T cell responses and antibodies in patients at the two ends of the spectrum. Tuberculoid leprosy at one end presents with localised hypopigmented paucibacillary skin patches, and shows effective antigen-specific T cell responses and low antibodies. In contrast, lepromatous leprosy at the other end presents with generalised lesions with bacillary proliferation, abundant antibodies, and T cell unresponsiveness/anergy to M. leprae. Recent advances that may explain clinical divergence and T cell unresponsiveness/anergy associated with lepromatous leprosy include: cytokine dysregulation, T helper (Th)1, Th2 paradigm, Th17 cells, FOXP3+ regulatory T cells, and pathogen-induced accessory cell subversion.
INTRODUCTION
Leprosy is a chronic infectious disease caused by Mycobacterium leprae which is non-cultivable by conventional methods. This ancient disease, seen less frequently in the developed world, is a public health concern in Asia1 and Latin America. The genome of the pathogen is known2 making it possible to now define antigens, examine their influence, and utilise specific antigens to dissect the immune basis of the disease, as well as develop diagnostics for early leprosy. Leprosy serves as a valuable model for gaining insights into human immune responses to infections, autoimmune disorders, and cancers.3 Based on the host immune response, it presents in diverse forms, from localised to generalised disease in different individuals. The clinical manifestations of the disease are mainly characterised by involvement of skin and peripheral nerves. Leprosy has been classified into five types and represents a spectrum based on the host immunity, wherein polar stable forms of localised paucibacillary dermal patches are seen in tuberculoid leprosy (T-lep), and generalised lesions with bacilli are seen in lepromatous leprosy (L-lep). In between these polar forms are the less stable forms of borderline T-lep, borderline L-lep, and borderline-borderline leprosy.4 Though many patients show a bland clinical course, 15–20% of patients develop leprosy reactions which are episodic, a cause of morbidity leading to peripheral nerve damage and requiring clinical intervention. They are classified into Type 1 or reversal reactions (RR) which present as inflammation of the local patches, mainly in borderline forms of leprosy and systemic Type 2 reactions or erythema nodosum leprosum (ENL), which present with fever, small reddish nodules scattered over the body, and joint pain in L-lep patients.5 T cells are key to the elimination of intracellular pathogens as well as malignant cells.3 Both escape T cell recognition by subverting innate and adaptive immune responses, thereby resulting in T cell unresponsiveness/anergy. This review will focus mainly on the immune basis of the divergence seen between T-lep and L-lep forms, in both stable and reactional states (Figure 1).
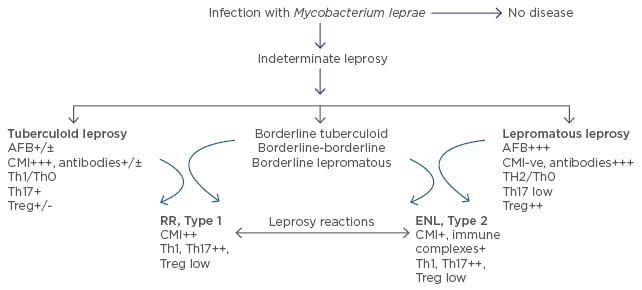
Figure 1: Clinicopathological classification of leprosy with significant immunological features under each leprosy type.
AFB: acid fast bacilli; CMI: cell mediated immunity; Th: T helper; Treg: T regulatory cells (FOXP3+); ENL: erythema nodosum leprosum; RR: reversal reactions.
The divergence between L-lep and T-lep patients lies in the former showing T cell unresponsiveness/anergy while mounting a good cytokine-dependent antibody response, whereas the latter shows the reverse pattern of immune responses. T cells are core to the protective immune response, evidenced by the existence of bacilli in L-lep patients. Moreover, T cell anergy is exquisitely antigen-specific and as such, patients mount responses to other bacteria including the related Mycobacterium tuberculosis. The antigen-specific anergy and the immunological basis for the clinical divergence associated with leprosy pose intriguing questions and have been investigated extensively in humans, as a suitable animal model is unavailable.6,7
DIVERGENCE IN ADAPTIVE IMMUNITY: T CELL RESPONSES
T-lep patients mount delayed-type hypersensitivity in skin tests and show ex vivo T cell responses to M. leprae antigens. Though integral or sonicated, M. leprae is not recognised by L-lep patients in skin tests or in ex vivo studies of peripheral blood cells;6,7 certain peptides/antigenic determinants of M. leprae stimulate the T cells of L-lep patients.6-9 Moreover, L-lep patients undergoing ENL reactions appear to recognise distinct peptides compared with the non-reaction group, both in cellular immune responses and antibody responses.6,8 These findings indicate that L-lep patients have T cells that recognise some antigenic sites of the pathogen which may have been cryptic, and such T cells may emerge during alterations in clinical state, as in reactions akin to epitope spreading.
CYTOKINE DYSREGULATION
The divergence of cell-mediated and antibody-mediated responses in leprosy was due to different T helper (Th) subsets with divergent cytokine release. T-lep patients were reported to have a Th1 profile with interferon-γ (IFN-γ) and interleukin (IL)-2, whereas a Th2 profile with IL-4 and IL-10 was associated with L-lep,6,7 thereby explaining the promotion of cell-mediated responses in the former and antibody responses in the latter. However, other studies including ours showed that this binary view may not provide a total explanation, as many T-lep and L-lep patients showed presence of a non-polarised Th0 subset of CD4+ cells releasing both IFN-γ and IL-4.10 It is of interest that during Type 2/ENL reactions, L-lep patients show a switch to a Th1 profile with IFN-g production and low/absent IL-4 indicating cytokine dysregulation, which may lead to tissue damage.11 That the Th switch may be associated with dendritic cells (DCs) was indicated by in vitro studies of peripheral blood mononuclear cells (PBMCs).12 Of clinical interest was the therapeutic potential shown by IFN-γ; L-lep patients given intramuscular IFN-γ showed rapid bacillary clearance in L-lep lesions compared with multidrug therapy alone.13
T HELPER 17 CELLS
Another CD4+ Th subset that shows divergence in leprosy is the Th17 population14 associated with inflammation. The Th17 subset is highest in healthy contacts exposed to the diseases compared with patients, indicating its importance in innate immunity.15,16 Moreover, T-lep patients showed higher expression of CD4+ Th17 cells than L-lep, both in stimulated PBMC and in situ skin lesions.16,17 Th17 was associated with its signature transcription factor RORC. Though transcription factor STAT3 was expressed in similar amounts in both types of leprosy subjects, the difference was in its phosphorylation status, with only T-lep showing phosphorylated STAT3. IL-23 and IL-23 receptors, known to be important for the differentiation of Th17 cells, also showed a similar divergence in the leprosy types. That the Th17 was an effector cell was indicated by its association with CCR6, a chemokine receptor. Th17 cells appeared in the leprosy patients who showed non-polarised Th0 subset.15 Therefore it would appear that Th17 plays a role in the immune responses to M. leprae infection and may be an alternate pathway for bacillary clearance both in the early and later stages of infection. The associated chemokines may help in its migration to lesional sites.
Both types of reactional leprosy patients showed an increase in Th17 cells in comparison to the non-reactional patients of the identical leprosy type, as attested by the presence of IL-17A and IL-17F in CD4+ cells.18 There was a difference between the two reactions, in which the associated cytokine IL-21 showed an increase in ENL subjects and not in RR/Type 1 reaction subjects. In general, Type 1 reactions, seen more with borderline T-lep, showed higher expression of cytokines, cytokine receptors, and chemokines compared with ENL. The inflammation noted in lesions of leprosy reactions is not only due to IFN-γ of the Th1 subset but is also contributed to by IL-17, the signature cytokine of the Th17 subset. IL-17 is also detectable in the serum of healthy contacts and T-lep patients and may be useful as a surrogate marker for monitoring treatment response19 and vaccine efficacy. IL-17 may also negatively regulate nerve growth factors and their receptors.20 As these growth factors affect peripheral nerves, IL-17 may consequently contribute to the nerve damage characteristically associated with this disease. It is not clear whether Th17 cells reflect T cell plasticity or constitute a stable lineage as most studies on patients are undertaken at a single point during an ongoing clinical course. Thus, whether it is a rescue pathway or alternate defence mechanism needs further investigation.
FOXP3+ T REGULATORY CELLS
Another distinct lineage of T cells that has exciting implications for dampening inflammatory responses is the T regulatory cell (Treg), which has a CD4+CD25+ nuclear FOXP3+ phenotype and shares a similar differentiation pathway to Th17 cells, although they have opposite effects. They were first identified in murine models of autoimmune diseases21 where they were shown to inhibit inflammatory responses. Since then, CD4+ Treg cells have been identified in humans,22 in autoimmune diseases,23 and in infectious diseases such as leishmaniasis24 and tuberculosis.25 Several types of Tregs have been described; some are natural Tregs derived from the thymus and act via contact with target cells.26 Others are inducible and mediate inhibition through cytokines such as transforming growth factor-β (TGF-β) and IL-10 (induced Treg [iTreg]).27 Transcription factor FOXP3 is thought to be the primary requirement for the suppressive function, though low and transient expression has been reported in activated human T cells with and without suppressor function. Though Tregs in mice express CD25 constitutively, in humans only those with CD25hi show suppressive function. Tregs revive the earlier concept of T suppressor cells in the 1970s and 1980s which was discarded when specific phenotypic markers could not be identified. The presence of T suppressor cells in L-lep patients was conflicting at that time. CD4+ or CD8+ populations of cells with inhibitory properties in ex vivo PBMCs derived from L-lep patients were reported by some28 and refuted by others.29
The discovery of FOXP3+ Tregs has revived the quest for a cellular basis for the anergy noted in leprosy. Tregs have been shown by several studies to be enhanced in L-lep patients both in PBMCs and in skin lesions.17,30-34 Though unstimulated PBMCs in our study also showed an increase in CD4+ Tregs, this was further enhanced by antigen stimulation, indicating recall responses. More importantly, TGF-β was associated only with CD4+ and not CD8+ cells. Furthermore, both CD25hi and CD25low cells showed divergence in the two types of leprosy with an increase in L-lep subjects. TGF-β required phosphorylation of the STAT5 transcription factor. Inflammatory cytokines IFN-γ or IL-17 were not detectable in CD25hi cells. It would appear therefore that Tregs play a significant role in L-lep and may downregulate antigen-specific T cell responses through TGF-β, and fall into the peripheral iTreg population. That TGF-β leads to facilitation and stability of FOXP3 through enhanced phosphorylation of SMAD3 and NFTAC was also shown in leprosy patients.32 Some studies have implicated the increased interaction of nuclear FOXP3 with histone deacetylase and transcription regulation for the immune suppression noted with these cells.33,34 This interaction downregulates gene expression of co-stimulatory CTLA-4, and may thereby inhibit T cell proliferation and cell-mediated immune responses to M. leprae. It has also been shown that macrophages infected with live M. leprae primed T cells towards FOXP3+ Treg cell differentiation, and inhibited Th1 cytokines and CD8+ cytotoxicity.35
Leprosy reactions in contrast showed a decrease in Treg cells which paralleled the increase in Th17 population.18 Moreover, there was downregulation of intracellular TGF-β.18,34 Most reports showed a reduction in Treg cells in patients with ENL reactions compared with non-reaction L-lep counterparts.18,34 However, reports of patients with RR differed on the presence of Treg activity.18,34 Recent evidence indicates that Th17 differentiation may be controlled by opposing roles of TGF-β and IL-6. Our studies indicated that monocyte-derived IL-6 may play a role in Th17 differentiation.18 Thus, the inflammation associated with Th1 and Th17 cells in leprosy reactions may be due to the lowering of the dampening effects of iTreg cells.
Of interest is the finding that healthy contacts exposed to the disease had higher iTregs than those with T-lep, suggesting that early dampening of the immune responses may have a protective role.18,35 However, this needs to be reconciled with the findings reported above of increased Th17 cells also seen in the healthy contacts of patients treated with chemotherapy.16
DIVERGENCE IN INNATE IMMUNITY AND SUBVERSION OF ACCESSORY CELLS BY MYCOBACTERIUM LEPRAE
Innate immunity is the first defence mechanism when a pathogen enters the host. Though there is overlap between the adaptive and innate immunity, several studies have shown differences in the latter which may contribute to the divergence in the clinical types of leprosy.36 Macrophages and DCs play an important role in the recognition of the pathogen during innate immunity37 as well as in triggering the adaptive immune system. The former has natural microbicidal ability that is further enhanced when primed by inflammatory cytokines such as IFN-γ and IL-17. M. leprae appears to subvert macrophage and Schwann cell functions in multiple ways. Foamy macrophages seen in skin lesions of L-lep inhibit bactericidal activity through phenolic glycolipid (PGE)2 of the pathogen, and have lipids that may provide a carbon source to promote mycobacterial proliferation.38 Analysis of the M. leprae proteome shows the presence of fatty acid β-oxidation enzymes.38 That lipid antigen may play a role was indicated in paucibacillary skin lesions of T-lep patients which showed enhanced CD1 proteins on CD83+ DCs, whereas the bacilli-laden L-lep lesions showed an absence.39
Macrophages have opposing functions of both killing and promoting M. leprae proliferation in the two types of leprosy.36,37 The M1 type of macrophages are pro-inflammatory and promote Th1 cytokine IFN-γ. On the other hand, M2 macrophages are anti-inflammatory, being associated with the Th2 cytokines IL-4, IL-10, and IL-13. L-lep monocytes were shown to be inhibitory for in vitro lymphoproliferation29 through the release of factors such as PGE2, leukotrienes, and IL-10.6 Live M. leprae infected macrophages of M2 type influenced Treg polarisation.40 IL-10 and IL-15 are innate immune cytokines seen in leprosy lesions, with the former being associated with L-lep and the latter with T-lep. Though both cytokines enhance CD209 (C-type lectin) expression on monocytes, IL-10 promotes phagocytosis whereas IL-15 induces the vitamin D-dependent microbicidal pathway. The former pathway was prominent in the L-lep and the latter in the T-lep form of the disease.37
DCs are also key players in antigen presentation and play a major role in leprosy.36,39 L-lep lesions have reduced DCs in skin lesions. B7-1, a co-stimulatory molecule, is reduced in L-lep lesions,41 and peripheral monocytes from L-lep patients fail to differentiate into CD1+ DCs. It has been suggested that M. leprae interferes with DC differentiation, promotes host-derived oxidised phospholipids seen in the L-lep lesions,38,42 and thus affects antigen presentation for effective T cell immunity.
Schwann cells are also host cells for M. leprae and appear to influence both innate and adaptive immune responses in leprosy. They have been implicated in the demyelination of peripheral nerves which is associated with this disease in the absence of immune cells,43 by interacting with the PGE1 of M. leprae. Recent studies in a murine model have shown that M. leprae reprogrammes Schwann cells to a stage where they become migratory and promote the spread of infection directly or through recruited macrophages by releasing chemokines, cytokines, and growth factors.44 Schwann cells have been shown to express CD209 and toll-like receptors (TLRs) in nerves of L-lep. Using Schwann cell lines, it was shown that CD209 and M. leprae binding was increased by Th2 cytokines IL-4 and IL-10, and not Th1 cytokine IFN-γ.45
Macrophages identify pathogens through pattern recognition receptors (PRRs) that recognise pathogen-associated molecular patterns present on the organisms. TLRs are PRRs on accessory cells which have been shown to interact with lipoproteins of mycobacteria and trigger host immune responses.46 TLR2-TLR1 heterodimers lead to activation of macrophages, DCs, and M. leprae death.47,48 They have also been observed in the Schwann cells of peripheral nerves. T-lep lesions show stronger expression of TLR1 and TLR2 compared to L-lep lesions.48 Macrophage differentiation to DCs via NOD2 was dependent on IL-32, and showed improved antigen presentation to CD8+ T cells.49 Moreover, increased expression of nucleotide-binding oligomerisation domain containing protein 2 (NOD2) and IL-32 was seen in T-lep skin lesions. Major membrane protein II of the pathogen, a lipoprotein, triggered macrophages and DCs through TLR2;47,50 such activation was enhanced by Th1 and inhibited by Th2 cytokines. Activation of TLR2/1 led to the production of inflammatory cytokine tumour necrosis factor-α as well as IL-12, promoting Th1 polarisation in the adaptive immune response.48 TLR activation also results in granulocyte macrophage colony-stimulating factor (GM-CSF) release that expands DCs, releasing IL-15 which promotes macrophage differentiation.48 TLR function in leprosy is regulated by cytokines. IFN-γ enhanced TLR1 expression, whereas IL-4 reduced TLR2 expression as well as cytokine release.48 IL-10 on the other hand did not affect TLR2/1 expression but inhibited cytokine release. Phospholipids inhibited TLR2/1-induced IL-12, but not IL-10. TLR1, TLR2, and TLR4 have identified single nucleotide polymorphisms associated with the pathogenesis of leprosy, including protection against leprosy and Type 1 leprosy reactions.51-53 Association of genes coding for host responses were reviewed recently.53,54 DNA sensing via TLR9 was involved in ENL, in which the E6446 synthetic TLR9 antagonist inhibited pro-inflammatory cytokines.55
The antimicrobial effect of TLR engagement is independent of nitric oxide50 and has been attributed to IL-15-dependent activation of vitamin D receptors with the induction of antimicrobial peptide cathelicidin.37,55,56 Genes encoding the vitamin D pathway were differentially expressed in T-lep and L-lep lesions.56 Moreover, miRNAs which have been shown to influence T cell differentiation, and activity of accessory cells has also been observed in leprosy. miRNA-21 seen in L-lep was shown to downregulate TLR2/1, upregulate IL-10, and inhibit vitamin D-dependent antimicrobial peptides.57 M. leprae also upregulates expression of tryptophan aspartate coat protein (TACO) in macrophages58 and downregulates TLR2-mediated signalling.59 TACO has been shown in leprosy lesions as well as in M. leprae containing macrophages in vitro.
Another intracellular PRR present in macrophages is NOD2. Mycobacterial ligand binds to NOD2, resulting in activation and rapid differentiation of monocytes to CD1+ DCs and requires IL-32. Such DCs are more efficient for priming CD8+ T cells than the GM-CSF derived DCs. IL-32-secreting DCs are more prevalent among T-lep than L-lep patients.37,49
CONCLUSION
In summary, divergence in both innate and adaptive immune responses is notably distinct in restricted, compared to generalised, forms of leprosy. The innate immune cells may promote resistance or susceptibility to the disease, not only by confronting the pathogen but also by instructing an appropriate adaptive immune response. M. leprae escapes being killed by evading both types of host response in L-lep patients. The way in which it avoids or masks recognition, or promotes T cell plasticity to manoeuvre the immune response towards anergy, provides insights for understanding the pathogenesis of other infectious diseases as well as autoimmunity and cancers.





