Summary
The over-production of TNF-α can lead to chronic inflammation and organ damage in immune-mediated inflammatory diseases (IMID), such as rheumatoid arthritis (RA), axial spondyloarthritis, psoriasis, and inflammatory bowel disease (IBD). Anti-TNF therapy is generally considered to be an effective, well-tolerated treatment option for the management of chronic inflammation in these conditions.
Over the past decade, patents for the original reference anti-TNF agents have expired, permitting the development of anti-TNF products that are biologically similar, termed ‘biosimilar’, to the original reference product. Differences in the approval process mean that biosimilars are often available to healthcare services at a considerably lower cost compared with the reference products, providing an opportunity to improve patient access to the benefits of anti-TNF therapy.
However, despite the spreading use of biosimilars across healthcare services, some clinicians remain reluctant to prescribe them. The gradual accumulation of long-term data on the real-world use of biosimilars, and an improved understanding of the development and approval process for these products, may help to increase clinicians’ confidence to increase usage of biosimilars.
This mini review summarises the current status of anti-TNF biosimilars in clinical practice, including the requirements for regulatory approval, real-word evidence for their equivalence to novel anti-TNFs, guidelines for their use, and challenges to their acceptance by both clinicians and patients.
INTRODUCTION
IMIDs such as RA, axial spondyloarthritis, psoriasis, and IBD affect 5–7% of the Western population.1,2 There is an increasing understanding of the overlapping pathogenesis between IMIDs, and today’s treatments focus on the pathological mechanisms behind these diseases.2,3
The advent of anti-TNF inhibitors in the late 1990s revolutionised treatment for patients with IMID, and remains the cornerstone for their treatment today.4,5 To date, there are five novel anti-TNFs currently approved for use in IMIDs: adalimumab, infliximab, golimumab, etanercept, and certolizumab pegol.6-11 However, since the patent that granted marketing exclusivity to infliximab expired in 2013, and patents for other anti-TNFs have followed, it has been possible to commercialise products that are biosimilar to these reference products.12
This mini review summarises the current status of anti-TNF biosimilars in clinical practice, including the requirements for regulatory approval, real-word evidence for their equivalence to novel anti-TNFs, guidelines for their use, and challenges to their acceptance by both clinicians and patients.
WHAT IS A BIOSIMILAR?
The European Medicines Agency (EMA) defines a biosimilar as “a biological medicinal product that contains a version of the active substance of an already authorised original biological medicinal product (reference medicinal product) in the European Economic Area [EEA].”13
Unlike ‘generic’ medicines, which can be chemically synthesised to be identical to reference molecule products, the reference products for biosimilars are large and/or complex molecules (biologics) that require cells for their manufacture.14 Although the genetic sequence can be pre-specified, the final product may be modified by the cell in different ways, introducing heterogeneity even between batches of the reference product.15
Anti-TNF Biosimilars Approved in Europe
The first anti-TNF biosimilar to be approved by the EMA was CT-P13, a biosimilar of infliximab,12 marketed by two pharmaceutical companies as Remsima and Inflectra. Over the past decade, the EMA has approved 21 anti-TNF biosimilars, 14 of which are based on adalimumab, four on infliximab, and three on etanercept (Table 1).16,17
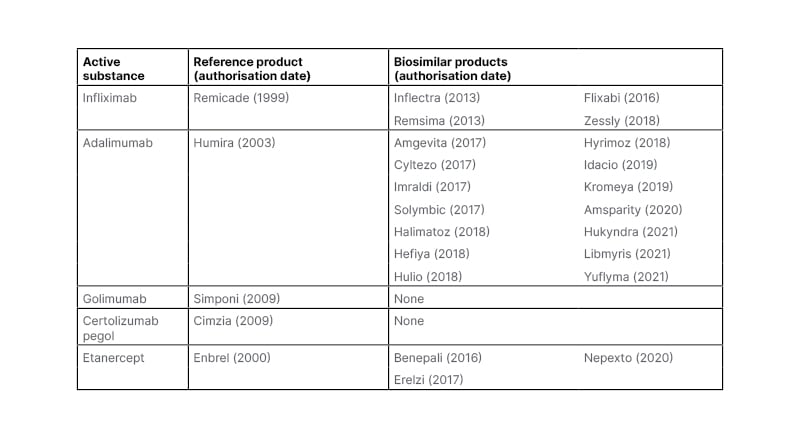
Table 1: Anti-TNF inhibitors approved by the European Medicines Agency (EMA) as of September 2022.
Biosimilars have grown to represent a substantial portion of the anti-TNF market in Europe, with adalimumab and etanercept reaching 53% and 54%, respectively, of the market share with their reference products in 2021.17 Switching between reference products and biosimilars is relatively common; in Italy, 46% of patients taking anti-TNF biosimilars between 2015 and 2019 switched from reference and biosimilar medicines, or vice versa.18
Benefits of Biosimilars
The development of biosimilars is less costly than that of the reference product as drug discovery research is not required; therefore, they can be made available to patients at a lower cost than the reference product.15 In addition, the existence of multiple products with the same ‘active substance’ can introduce competition into the market, generally resulting in a price-lowering effect.15 This economic benefit, combined with updates to treatment guidelines and widespread reimbursement policies, has increased patient access to anti-TNF therapy across Europe.19
The impact of anti-TNF biosimilars may increase opportunities to realise clinical goals in IMID, such as early and longer biological treatment, treating to target, reducing disease burden, and maintenance of remission.20
It has also been suggested that advances in molecular engineering in the past decade might permit the development of anti-TNF biosimilars that improve upon the properties of the reference product.4 For example, while the reference product for infliximab, Remicade, does not have a subcutaneous formulation, biosimilar Remsima has been developed and approved to be used in this way.21
REGULATORY REQUIREMENTS FOR THE APPROVAL OF BIOSIMILARS
The same standards of quality, safety, and efficacy that cover all biological medicines approved in the European Union (EU) are also applied to biosimilars.22 Licencing of biosimilars by the EMA is subject to strict regulatory control.23
However, when assessing medicines for marketing approval, different questions apply to biosimilars than to the original reference product. Rather than demonstrating the effect of the medicine and its mechanism of action, drug developers need to demonstrate biosimilarity to the reference product; that is, a highly similar structure and function, as well as safety and immunogenicity profiles.15,22
The complex analytical characterisation of a biosimilar and comparison to a reference product (Figure 1) is far more sensitive in detecting any differences than a clinical trial could be.15 For this reason, the emphasis for approving a biosimilar is far more focused on the pre-clinical studies than a novel reference product would be.15 Nevertheless, there remains a clinical aspect to biosimilar development as comparative clinical trials are conducted to confirm biosimilarity.22
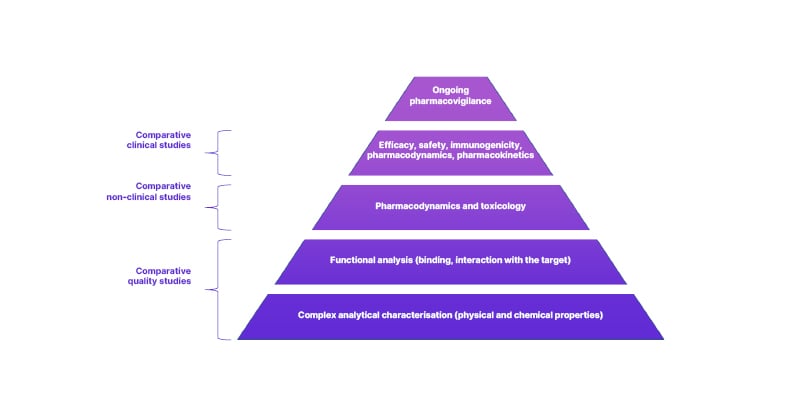
Figure 1: Biosimilar assessment is based on comparative studies, founded on physical, chemical, and functional analyses.
Adapted from European Medical Journal (EMJ, 2022)16 and European Medicines Agency (EMA, 2019).23
At the clinical trial level, a biosimilar is only required to demonstrate comparable safety and efficacy in one therapeutic indication. The safety and efficacy in other indications can be extrapolated from the reference product, avoiding unnecessary repetition of clinical trials.15,22 Paul Cornes, a consultant oncologist in Bristol, UK, and core lecturer at the European School of Oncology (ESO), explained that comparative trials are not always performed in what might be considered the major indication for the medicine, but the one most sensitive to detect any difference.15
Because of the limited opportunity for very rare safety events to be detected during the minimal clinical trials required for biosimilars, post-marketing safety monitoring is a formal regulatory requirement for these products in the EU.23 Furthermore, ongoing pharmacovigilance is necessary to capture the immunogenicity of biosimilars which, like their reference products, have the potential to induce an immune response which may result in adverse reactions and loss of efficacy.24
REAL-WORLD STUDIES OF ANTI-TNF BIOSIMILARS IN IMMUNE-MEDIATED INFLAMMATORY DISEASES
Since the first biosimilars were approved, many studies have assessed the safety and efficacy of switching from the reference products.25 A systematic review of 90 studies in 2018 reported that the vast majority of studies did not observe differences in safety, efficacy, or immunogenicity between biosimilars and the associated reference products, indicating that the benefit–risk profile is unchanged when switching to a biosimilar.26
Some of the most recent real-world studies conducted in this field, published over the past 18 months, are summarised below.
An observational study in patients switching from infliximab to an infliximab biosimilar for the treatment of IMID (n=48) showed that there were no significant differences in clinicians’ perception of disease activity, patient-reported outcomes, or laboratory parameters over 2 years following the switch. However, nearly a quarter of patients (12/48) experienced either disease relapse or serious adverse events (41% of patients with sarcoidosis), leading authors to recommend careful consideration prior to switching, particularly in patients with granulomatous diseases, such as sarcoidosis.27
In psoriasis, anti-TNF biosimilars have been shown to be effective and well-tolerated in adults (n=73),28 children (n=11),29 and the elderly (n=23),30 with no statistically significant differences observed between the biosimilars and their reference products.
An Italian study in patients with IBD (n=156) who were treated with adalimumab or one of two biosimilars found no statistically significant differences in clinical benefit between groups after induction or at 6 months, and all treatments demonstrated a good safety profile.30 Positive efficacy and safety outcomes have also been reported with the use of an infliximab biosimilar in children with IBD (n=87).31 Real-world data from Italy,32 France,33 the Netherlands,34 Poland,35 and the USA36 published on switching from infliximab to an infliximab biosimilar in patients with IBD reported that biosimilars had a similar efficacy, safety, and immunogenicity profile to that of the reference product.32-36
One study showed that 9.9% (75/758) of patients with IBD ‘reverse switched’ from a biosimilar back to the reference product.34 The most commonly reported reasons for reverse switching were gastrointestinal symptoms (25.5%) and dermatological symptoms (21.8%), with 12.0% of patients reverse switching due to loss of treatment response.34 Reverse switching led to an improvement in reported symptoms in 73.3% of patients, and seven out of nine patients (77.8%) with loss of response regained response.34
Several real-world studies of biosimilars in inflammatory rheumatic joint diseases have also recently been published, all indicating that these treatments were effective and well-tolerated.37-41
In addition, small studies of infliximab and adalimumab biosimilars have indicated promising efficacy and safety in patients with Takayasu arteritis (TAKASIM; n=23), a systemic vasculitis affecting the aorta and its branches, and Behçet’s uveitis (n=48), respectively.42,43
An analysis of long-term safety data for biosimilars in the EU was conducted in 2021. Results indicated that post-marketing surveillance data for biosimilar monoclonal antibodies and fusion proteins, covering up to 7 years of follow-up, did not reveal any adverse events specific to biosimilars.44
WHAT DO THE GUIDELINES SAY?
In an educational webinar, ‘Biosimilars within Haematology & Oncology’ held in 2022, Arnold Vulto, Emeritus Professor of Pharmacy at Erasmus University Medical Centre, Rotterdam, the Netherlands, suggested that a lack of clear guidance is undermining clinicians’ trust in biosimilars. Vulto emphasised that although the EMA and U.S. Food and Drug Administration (FDA) are excellent sources of regulatory information for biosimilars, their websites contain a vast amount of data, and this means that local clinicians often prefer to look for information from their local regulatory agency. Vulto stated that unfortunately, this information is not available in many European countries.15
The European Specialist Nursing Organisation (ESNO) explains that the choice to switch from a reference product to a biosimilar is made by the clinical decision maker, and their options can vary according to national and local policies. Although in Europe the EMA approves a biosimilar medicine based on equivalence of the efficacy and safety to the reference product, local policy regarding interchangeability between these products is set by the national authorities.45
European treatment guidelines that specifically mention the use of anti-TNF biosimilars are largely supportive of their use in IMIDs.23,46 For example, the European Crohn’s Colitis Organisation (ECCO) 2017 position statement on the use of biosimilars for IBD emphasises ‘the decision to initiate a biologic, biosimilar, or non-medical biosimilar switch, should always consider patient preference. The information offered must be transparent and the requirement of a non-medical switch must be made clear to the patient.’23
ECCO statements include:
- Biosimilarity is more sensitively characterised by performing suitable in vitro assays than through clinical studies.
- Clinical studies of equivalence in the most sensitive indication can provide the basis for extrapolation.
- When a biosimilar product is registered in the EU, it is considered to be as efficacious as the reference product when used in accordance with the information provided in the summary of product characteristics.
- Demonstration of safety of biosimilars requires large observational studies with long-term follow-up in patients with IBD.
- Adverse events and loss of response due to immunogenicity to a biologic drug cannot be expected to be overcome with a biosimilar of the same molecule.
- Switching from the originator to a biosimilar in patients with IBD is acceptable.
- Switching from originator to a biosimilar should be performed following appropriate discussion between clinicians, nurses, pharmacists, and patients, and according to national recommendations.
The European League Against Rheumatism (EULAR) 2019 recommendations for the management of RA equates reference anti-TNFs with EMA- or FDA-approved biosimilars.47 They stress that if therapy with an anti-TNF has failed, this should not be replaced with a biosimilar of the failed compound.47 Similarly, the European Guidelines in Dermatology (EuroGuiDerm) guideline on psoriasis emphasises that their recommendations apply equally to the reference medicine and biosimilars.48
CHALLENGES IN THE ACCEPTANCE OF ANTI-TNF BIOSIMILARS
Liese Barbier, a researcher at the University of the Regulatory Sciences and Pharmacoeconomics Research Unit, Katholieke Universiteit (KU) Leuven, Belgium, explained that although the EMA performs a centralised assessment for all biosimilars, many national regulators do not follow the same approaches to assessments, and this leads to differences in the information and guidance they provide.15,49
An online patient survey conducted in 2014–2015 indicated that of the 1,181 patients that responded, only 38% had heard of biosimilars. Nearly half of respondents (47%) were concerned about the safety profile of biosimilars, with 40% and 35%, respectively, expressing concern about efficacy and molecular basis.50 This survey highlights the challenges in informing and involving patients in shared decision making.
A similar survey of specialist physicians who are high prescribers of biologics (n=1,201), conducted between 2015–2016, highlighted a need for evidence-based education about biosimilars in this cohort. For example, knowledge of the fundamentals of biosimilars, and of the concept of extrapolation, was low, and less than half of respondents stated that biosimilars were safe and appropriate for use in naïve and existing patients.51 Reassuringly, a more recent survey of rheumatologists in the USA (n=320) in 2019 found that nearly all respondents were familiar with the FDA definition of a biosimilar product, and that they had a good understanding and acceptance of anti-TNF biosimilars.52
However, many clinicians remain reluctant to use biosimilars, particularly if this requires switching their patients from a reference anti-TNF.15 This may be partly due to the fact that the biosimilar development model, and the science behind it, are outside of the field of expertise of most clinicians. As a result, reasons for the reluctance to prescribe biosimilars include the use of the term ‘similar’, suggesting that the medicine is fundamentally not the same as the reference medicine, and the fact that clinical trials form a confirmatory, rather than a core, aspect of the assessment of biosimilars.53 Cornes explained that physicians may be hesitant to prescribe a biosimilar, or any medication they are not confident of, because the initial course of treatment is critical in patients with more severe disease.15
Although studies have generally reported similar efficacy and safety between anti-TNFs and biosimilars, some have reported an increased risk of treatment failures or adverse events in patients who switch to a biosimilar, compared with those who remain on the reference medication.54 One potential reason for treatment failure is the nocebo effect, resulting from a patient’s negative expectations towards a change in therapy. Effective patient education and healthcare provider–patient communication is crucial to minimise any nocebo effects associated with switching to a biosimilar.55 However, patients who lose treatment response following a switch should be permitted to return to the reference product.54 Another potential factor is the buffer in which the medication is delivered, which can vary between the reference product and biosimilars, and which may be associated with injection site pain in some patients.56
CONCLUSIONS
The literature to date supports the use of anti-TNF biosimilars as safe and efficacious alternatives to their reference products. It is critical to increase the acceptance of anti-TNF biosimilars across healthcare stakeholders to improve patient access to these treatments.15,20
The gradual accumulation and analysis of long-term data on the use of biosimilars may help to increase the confidence of clinicians to advocate their use.25,57 It is also important for clinicians to understand the development and approval process for biosimilars in order to provide the best cost-effective access to care for their patients.25,57 For example, clinicians should understand that biosimilar products are approved on their ‘sameness’ to a reference product, not through the recreation of trials performed for the reference product. This sameness is performed in the setting most sensitive to detect any differences, which may not be the setting for the major indication of the product.15
When administering biosimilar anti-TNFs to patients, it is important to maintain good lines of communication between the clinician and patient in order to minimise nocebo effects and mitigate any adverse effects that might emerge.54,56 Finally, if a patient does experience a loss of efficacy or the emergence of adverse events following a switch to a biosimilar, they should be offered the option of returning to the reference product.54,56
The introduction of biosimilars can expand access to treatment for patients with IMIDs across Europe and potentially improve the financial burden of healthcare systems. As real-world evidence data grows, physician and patient acceptance should follow, not only to initiate treatment with a biosimilar but also to switch from a reference product to a biosimilar.